Translate this page into:
A novel frameshift mutation of the NF1 gene in a Chinese pedigree with neurofibromatosis type 1
Correspondence Address:
Shi Lian
Department of Dermatology and Venereology, Capital Medical University, 10 Youanmen St, Fengtai District, Beijing 100069
China
Wei Zhu
Department of Dermatology, Xuanwu Hospital, Capital Medical University, 45 Changchun St., Xuanwu District, Beijing 100053
China
| How to cite this article: Lin X, Chen H, Zhu W, Lian S. A novel frameshift mutation of the NF1 gene in a Chinese pedigree with neurofibromatosis type 1. Indian J Dermatol Venereol Leprol 2017;83:231-233 |
Sir,
Neurofibromatoses are a group of hereditary disorders, characterized by an autosomal dominant pattern of inheritance. The most common variant is neurofibromatosis type 1 (90%), which affects one in every 3500 individuals. Neurofibroma is the cutaneous hallmark of neurofibromatosis type 1. This tumor is often accompanied by café-au-lait macules, axillary freckling, Lisch nodules of the iris and bone lesions and these patients are predisposed to develop malignancies. Neurofibromatosis type 1 is caused by mutation of the NF1 gene which is located on chromosome 17q11.2 and contains sixty exons; it also spans more than 300 kb of genomic deoxyribonucleic acid.[1] Neurofibromin, which is encoded by NF1 gene is a large protein (with 2818 amino acids) expressed in all cells and is particularly expressed at a high level in neurons, Schwann cells, glial cells and leukocytes. NF1 gene mutation leads to haploinsufficiency of neurofibromin thereby resulting in increased risk of benign or malignant neoplasms.[2]
Two patients in one pedigree were clinically diagnosed with neurofibromatosis type 1 at Xuanwu Hospital, Capital Medical University, China. The proband was a 17-year-old boy born with four large café-au-lait macules on his waist and buttocks. He gradually developed other café-au-lait macules on his trunk which increased in size from 1 cm × 2 cm to 23 cm × 14 cm. At the age of 10, a tender mass was found on the dorsum of his left foot. This mass was resected twice, but recurred. The recurrent mass had a diameter of 4 cm [Figure - 1]. Histopathological features of the lesion were consistent with neurofibroma.
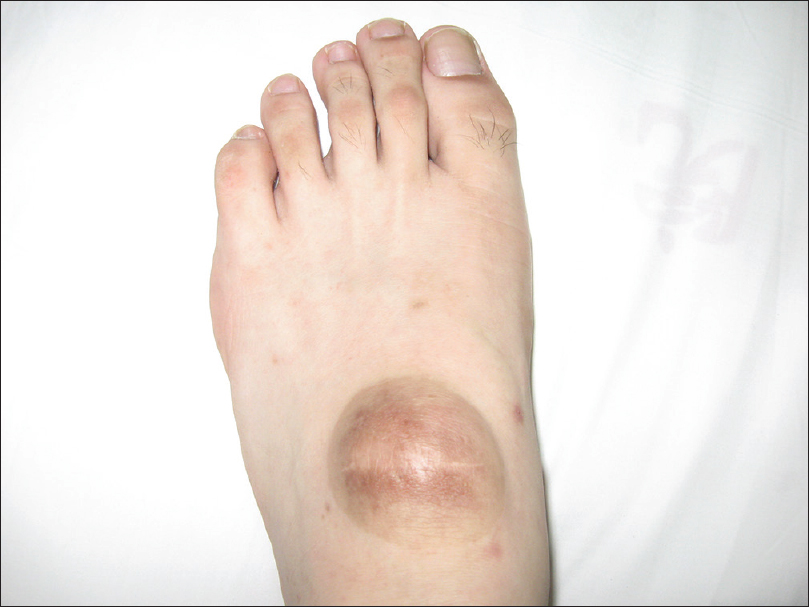 |
| Figure 1: Neurofibroma seen on the dorsum of the left foot in the proband |
The second patient was the boy's mother who was 53 years old. She was born with a large café-au-lait macule on her back. With age, she developed multiple subcutaneous neurofibromas and an increased number of café-au-lait macules all over her body [Figure - 2]. She had had mild dyspnoea and chest pain 40 years back, which on evaluation, was attributed to a mediastinal mass compressing the left lung. Histopathology of the resected tumour revealed a neurofibroma. No abnormality was found on ophthalmologic examination or magnetic resonance imaging (MRI) of the central nervous system.
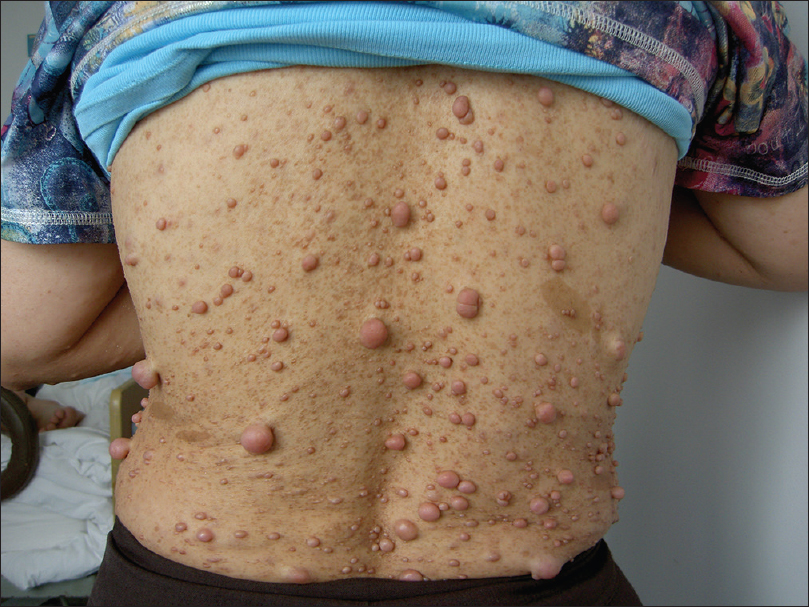 |
| Figure 2: Multiple caf�-au-lait macules and neurofibromas on the mother�s back |
This study was approved by the Ethical Committee of Xuanwu Hospital, Capital Medical University. We collected 5 ml of peripheral blood from each patient for genomic deoxyribonucleic acid extraction. All exons and exon/intron boundaries of the NF1 gene were amplified by polymerase chain reaction. Deoxyribonucleic acid samples from 100 controls with a normal phenotype were screened for novel NF1 mutations to exclude the possibility of polymorphism. After comparing the sequence with the reference complementary deoxyribonucleic acid (accession number NM_000267.3 in GenBank), we identified the mutations. A novel heterozygous frameshift mutation (c. 3236_3240dupTTCTA) in exon 25 of the NF1 gene mutation was identified in both the boy and his mother [Figure - 3]. This germline mutation produced a premature stop codon and resulted in a truncated protein instead of the full-length neurofibromin. No mutants were identified among the 100 normal control subjects. Hence, this novel germline mutation was probably the disease-causing mutation.
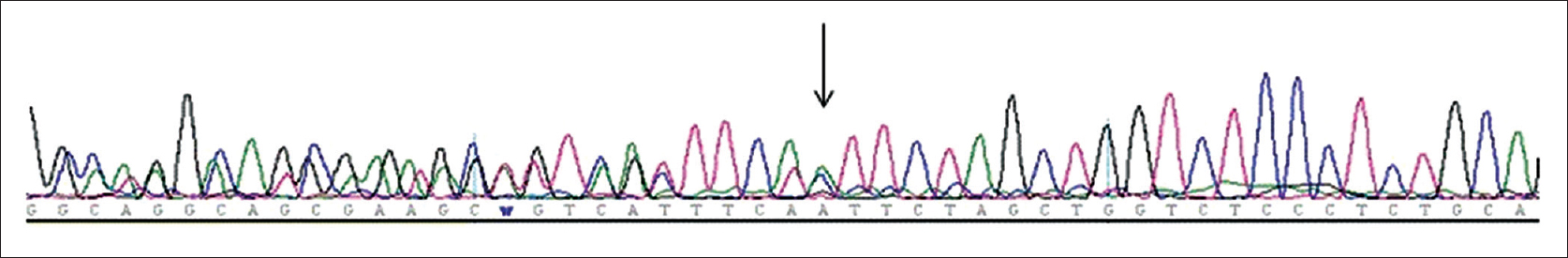 |
| Figure 3: Frameshift mutation c. 3236_3240dupTTCTA of partial deoxyribonucleic acid sequence of the NF1 gene. The mother and the son had the same mutation |
Neurofibromatosis type 1 is a complex hereditary neurocutaneous syndrome caused by mutations in the NF1 gene, one of the largest genes in the human genome. The NF1 gene has significantly high mutation rates caused by the large number of coding exons and mutational heterogeneity. To date, more than 1000 mutations of several types have been reported including chromosomal abnormalities, insertions, point mutations, deletions, stop mutations, 3'-untranslated region mutations and splicing mutations.[3] The novel frameshift mutation we detected significantly affected the function of the NF1 gene and resulted in an early termination of protein synthesis. In eukaryotes, nonsense-mediated decay is a surveillance mechanism observed for premature termination of translation. In this pathway, premature termination promotes the recruitment of a set of factors that degrade messenger ribonucleic acids undergoing this process.[4] As a result, the truncated proteins would not be synthesised. Both these factors may have played a role in the occurrence of the disease. The limitation of our study is that the truncated neurofibromin was not verified on a protein level; hence it is not fully proved whether the disease is caused by the frameshift mutation. The clinical presentation of neurofibromatosis type 1 is highly heterogeneous, ranging from the cutaneous manifestations alone to a number of medical complications. In this study, two patients had the same genotype but their phenotypes were different. It has been suggested that feature-specific modifier genes unlinked to the neurofibromatosis type 1 locus, epigenetic alterations or environmental factors may contribute to such variable expression.[5]
In conclusion, the c. 3236_3240dupTTCTA mutation in NF1 gene appears to be the cause of neurofibromatosis type 1 in this pedigree. This study adds a novel mutation to the spectrum of NF1 mutations and provides probable evidence that the loss or diminished function of the neurofibromin leads to neurofibromatosis type 1.
Acknowledgments
The authors sincerely thank the proband and his family members for participation in this study and also all the other participants for their consent to this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Ars E, Kruyer H, Morell M, Pros E, Serra E, Ravella A, et al. Recurrent mutations in the NF1 gene are common among neurofibromatosis type 1 patients. J Med Genet 2003;40:e82.
[Google Scholar]
|
| 2. |
Alkindy A, Chuzhanova N, Kini U, Cooper DN, Upadhyaya M. Genotype-phenotype associations in neurofibromatosis type 1 (NF1): An increased risk of tumor complications in patients with NF1 splice-site mutations? Hum Genomics 2012;6:12.
[Google Scholar]
|
| 3. |
Bottillo I, Ahlquist T, Brekke H, Danielsen SA, van den Berg E, Mertens F, et al. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J Pathol 2009;217:693-701.
[Google Scholar]
|
| 4. |
Kervestin S, Jacobson A. NMD: A multifaceted response to premature translational termination. Nat Rev Mol Cell Biol 2012;13:700-12.
[Google Scholar]
|
| 5. |
Sabbagh A, Pasmant E, Laurendeau I, Parfait B, Barbarot S, Guillot B, et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum Mol Genet 2009;18:2768-78.
[Google Scholar]
|
Fulltext Views
2,153
PDF downloads
1,103





![[Figure - 1]](#fig_ijdvl_2017_83_2_231_198457_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2017_83_2_231_198457_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2017_83_2_231_198457_f3.jpg){kind=link}