Translate this page into:
Drug hypersensitivity syndrome
Correspondence Address:
Devinder Mohan Thappa
Department of Dermatology and STD, Jawaharlal Institute of Postgraduate Medical Education and Research, Pondicherry - 605 006
India
| How to cite this article: Kumari R, Timshina DK, Thappa DM. Drug hypersensitivity syndrome. Indian J Dermatol Venereol Leprol 2011;77:7-15 |
Abstract
Drug hypersensitivity syndrome (DHS) is an adverse drug reaction commonly associated with the aromatic antiepileptic drugs (AEDs), viz., phenytoin (PHT), carbamazepine (CBZ), phenobarbital (PB), lamotrigine, primidone, etc. It can also be caused by other drugs, such as sulfonamides, dapsone, minocycline, gold derivatives, cyclosporine, captopril, diltiazem, terbinafine, azathioprine and allopurinol. Diagnosis of DHS may be difficult because of the variety of clinical and laboratory abnormalities and manifestations and because the syndrome may mimic infectious, neoplastic or collagen vascular disorders. The risk for developing hypersensitivity within 60 days of the first or second prescription in new users of PHT or CBZ was estimated to be 2.3-4.5 per 10,000 and 1-4.1 per 10,000, respectively. The syndrome is defined by the fever, skin rash, lymphadenopathy and internal organ involvement within the first 2-8 weeks after initiation of therapy. Internal manifestations include, among others, agranulocytosis, hepatitis, nephritis and myositis. Insufficient detoxification may lead to cell death or contribute to the formation of antigen that triggers an immune reaction. Cross-reactivity among PHT, CBZ and PB is as high as 70%-80%. Management mainly includes immediate withdrawal of the culprit drug, symptomatic treatment and systemic steroids or immunoglobulins.Introduction
Drug hypersensitivity syndrome (DHS), recently being also referred to as DRESS (drug reaction with eosinophilia and systemic symptoms) or DIDMOHS (drug-induced delayed multi-organ hypersensitivity syndrome), is increasingly being recognized as a distinct type of adverse drug reaction. [1],[2] It was first associated with the aromatic antiepileptic drugs (AEDs), viz., phenytoin (PHT), carbamazepine (CBZ), phenobarbital (PB), lamotrigine and primidone
(PRM). [3] The syndrome can also be caused by a variety of other drugs, such as sulfonamides, [4] dapsone, [5] minocycline, [6] terbinafine, [7] azathioprine, [8] allopurinol, [9] gold derivatives, cyclosporine, captopril, diltiazem, felbamate, [10] metronidazole, nonsteroidal anti-inflammatory drugs like ibuprofen and the antiretrovirals like nevirapine and abacavir, [11] etc. It is usually defined by the triad of fever, skin rash and symptomatic or asymptomatic internal organ involvement. [10]
History
Soon after the introduction of hydantoins in the early 1930s, there were first reports of several different adverse drug reactions, including rashes, fever and eosinophilia. [12] In 1950, Chaiken et al,[13] described ′dilantin hypersensitivity,′ a systemic hypersensitivity reaction associated with PHT drug therapy. However, it took another 20 years before a consensus was reached that PHT could produce a characteristic ′hypersensitivity syndrome′ consisting of fever, cutaneous eruption, lymphadenopathy, peripheral leukocytosis and sometimes life-threatening hepatic necrosis. [12],[14] It was later realized that other aromatic AEDs, viz., CBZ and PB, could also induce the same syndrome.
Nomenclature
In 1988 the term ′anticonvulsant hypersensitivity syndrome′ was first used to label this specific entity. [3] ′Dilantin hypersensitivity reaction,′ ′phenytoin/ Dilantin syndrome,′ ′Kawasaki-like syndrome,′ ′mononucleosis-like syndrome,′ pseudolymphoma, febrile mucocutaneous syndrome, graft-versus-host-like illness, Kawasaki-like illness, hypersensitivity mimicking infectious mononucleosis-like illness, multisystem hypersensitivity reaction, and drug-induced aseptic meningitis are the other names used to refer to a similar constellation of symptoms. [12],[15] Since misdiagnosis or delayed diagnosis can result in death for want of a correct diagnosis, it is crucial to adopt a standardized nomenclature and definition of DHS. Recently, other acronyms being used for DHS include DRESS and DIDMOHS. [1],[2]
Epidemiology
The incidence of DHS is approximately 1 in 1,000 to 1 in 10,000 exposures. [15] In a recent record-linkage study, the risk for developing a hypersensitivity within 60 days of the first or second prescription in new users of PHT or CBZ was estimated to be 2.3-4.5 per 10,000 and 1-4.1 per 10,000, respectively. [16] Studies have shown 80% cross-reactivity between the anticonvulsants. A temporal relationship between medicine use and onset of the syndrome is the most important indicator of causality. [17]
Clinical Features
DHS is characterized by a constellation of symptoms involving various organs and organ systems, particularly the skin, liver and hematologic system being most commonly involved, with cutaneous changes being the most apparent. Correct diagnosis of DHS may be difficult because of the wide variety of possible clinical and laboratory abnormalities and manifestations and because the syndrome may mimic infectious, neoplastic or collagen vascular disorders. [18]The presence of the 2 criteria, as shown in [Table - 1], given by Bocquet et al, has high sensitivity (>95%) but weak specificity (<80%). [11]

Drug hypersensitivity syndrome usually occurs on first exposure to the associated medication, with a delayed onset. Reactions classically begin 1 week to 8 weeks after starting drug therapy. For PHT, the mean interval to onset is 17 to 21 days; and for CBZ, the onset is generally between 21 and 28 days. In previously sensitized individuals, anticonvulsant hypersensitivity syndrome may occur within 1 day on re-challenge. Anticonvulsant hypersensitivity syndrome has no relationship to dosage or serum concentration of anticonvulsants. [15]
The reaction usually starts with low- or high-grade fever, and over the next 1 to 2 days a cutaneous reaction, lymphadenopathy and pharyngitis may develop. This is followed by involvement of various internal organs, most commonly the liver, although hematologic, renal or pulmonary impairment may occur. [15]
Cutaneous features
A drug eruption occurs in approximately 90% of patients with anticonvulsant hypersensitivity syndrome, which may be exanthematic, erythrodermic (with or without pustules) or blistering suggestive of Stevens-Johnson syndrome (SJS) or toxic epidermal necrolysis (TEN). [15] The severity of cutaneous changes does not necessarily reflect the severity of internal organ involvement; therefore, careful assessment is necessary for patients with any drug-associated eruption accompanied by systemic symptoms. The skin rash is most commonly an exanthem with or without pruritus. In most cases, the cutaneous eruption starts as a macular erythema that often evolves into a red, symmetrical, pruritic, confluent, papular rash. Initially, the upper trunk and face are affected, with later involvement of the lower extremities. Rarely, generalized follicular pustules or more severe skin reactions, such as exfoliative dermatitis or erythroderma, erythema multiforme, Stevens-Johnson syndrome or toxic epidermal necrolysis, may occur. [15] The incidence of these severe skin reactions as part of DHS was found to be as high as 9% among 53 patients with DHS induced by PHT, CBZ or PB reported by Shear and Spielberg; [19] or 13% among 38 cases of PHT hypersensitivity reported by Haruda. [20] Re-exposure after a previous adverse reaction or with continued treatment with the anticonvulsant after hypersensitivity develops can cause more severe reaction. Vesicles and tight blisters may be induced by dermal edema. Because there is no necrosis in the epidermis, these blisters are different from those of TEN but can be confusing in the absence of pathological examination.
Angioedema (especially facial or periorbital swelling) may be a sign of a systemic and potentially severe reaction. Tender local (usually involving cervical nodes) or generalized lymphadenopathy (usually axillary, cervical and inguinal nodes) is another common feature of the initial presentation of DHS. Patients with DHS usually present with benign lymphoid hyperplasia and sometimes conjunctivitis. [10] Facial or periorbital edema helps in the diagnosis, because the typical erythematous, symmetric drug eruption often involves the body but spares the face. The edema may induce blistering. An edematous, follicular accentuation is characteristic. Sterile follicle-centered pustules may eventuate. [21] With resolution of rash, desquamation occurs.
Extracutaneous manifestations [Table - 2]
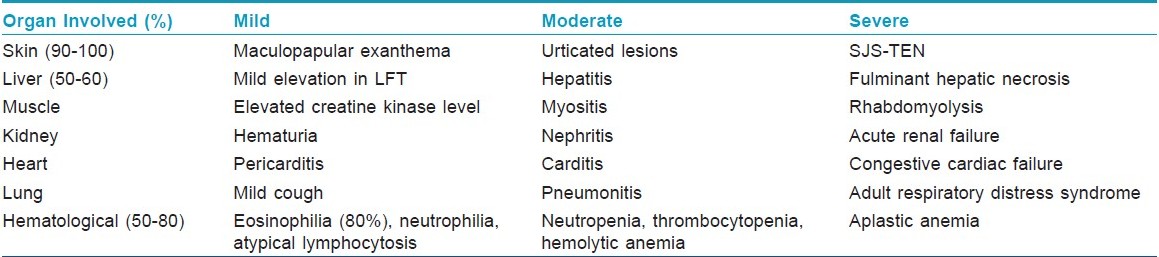
Fever (90%-100%) is usually high and spiking, ranging from 38ºC to 40ºC. The spikes in temperature may be suggestive of an underlying infection, but the results of cultures are negative. The spikes in temperature may persist for weeks after the offending drug is discontinued. This is especially true in those with a severe reaction.
Internal organ involvement may not develop for 1 to 2 weeks and may even develop 1 month later. [15] Various internal organs may be involved, particularly the liver, kidneys and hematologic system. Manifestations include hepatitis, nephritis, pneumonitis, neutrophilia, eosinophilia, atypical lymphocytosis, blood dyscrasias, hemolytic anemia and changes in immunoglobulin levels. [10],[15] Other findings include periorbital or facial edema (25%); oral ulceration, exudative tonsillitis, oral ulcers, strawberry tongue, hepatosplenomegaly, flu-like symptoms, myopathy, disseminated intravascular coagulopathy, pharyngitis (10%); hypotension, colitis, pancreatitis, myocarditis, myositis, meningitis, parotitis, orchitis, arthralgia (21%); and thyroiditis. Delayed-onset hypothyroidism has also been described. [21] Lymphadenopathy occurs in 75% of cases. [10],[12],[15]
The liver is the most frequently involved internal organ in DHS. The rate of liver involvement in patients with DHS has been reported to range from 34% to 94%. [20],[21],[22],[23] The presentation can range from mild elevations in transaminases to marked abnormalities in liver function tests with hepatomegaly to fulminant hepatic necrosis. [10] Severe hepatitis with jaundice increases the risk of mortality to between 12% and 50%. [15] The degree of hepatitis is related to the interval between the onset of the syndrome and the discontinuation of the anticonvulsant. This emphasizes the importance of prompt recognition of the syndrome. [18]
Blood abnormalities (50%) can present either as toxic (e.g., thrombocytopenia, neutropenia, agranulocytosis, Coombs-negative hemolytic anemia) or reactive (e.g., Coombs-positive hemolytic anemia, atypical lymphocytosis during the first week followed by striking eosinophilia) manifestations. [23] Rarely, leukopenia has been reported. The most characteristic biological alteration is eosinophilia, often above 1500/mm 3 , seen in 80% of cases. The eosinophilia may have a delayed onset of up to 1 to 2 weeks. An absolute eosinophil count of more than 1.5 x10 9 /L is toxic to endothelial cells and can lead to cardiac, gastrointestinal, central nervous system, pulmonary and renal dysfunction, including coronary artery thrombosis and eosinophilic pneumonitis. Mitotic forms may be present. A Coombs-negative hemolytic anemia can occur. Immunoglobulin, erythrocyte sedimentation rate, and complement levels are usually normal. Interstitial nephritis and severe rhabdomyolysis and myopathy have also been reported. In one case, there was a persistent panhypogammaglobulinemia.
Kidney involvement varies in severity from hematuria to nephritis to acute renal failure. Colitis may present as abdominal pain and diarrhea. [10]
Thyroid involvement has also been seen in a small subgroup of patients. The hyperthyroid phase of the illness, which develops during the acute phase of the reaction, may be missed by the clinician, because fever, tachycardia and malaise appear to be part of the anticonvulsant hypersensitivity syndrome. [10] These patients will then become hypothyroid as part of an autoimmune thyroiditis within 2 months of initiation of symptoms. It is characterized by low thyroxine level; an elevated level of thyroid-stimulating hormone and autoantibodies, including antimicrosomal antibodies. Resolution of the autoimmune thyroid disease associated with anticonvulsant hypersensitivity syndrome occurs over the ensuing 12 to 18 months, allowing eventual discontinuation of thyroid hormone replacement.
Course
The clinical course of DHS is variable. Initially, patients appear toxic. If DHS is recognized early and the culprit drug(s) is discontinued, the course is often less eventful, with the syndrome resolving over the next few weeks. The rash may disappear with mild desquamation. There is resolution of fever, and hematologic abnormalities become normal. Sometimes, however, even after discontinuation of the culprit AED, the hypersensitivity reaction process may progress, and the patient becomes worse, before any improvement is seen. The syndrome is sometimes fatal; particularly if hepatitis is present, the mortality ranges from 18% to 40%. Patients who are re-challenged with the culprit AED, whether inadvertently or in a controlled setting, develop the symptoms of fever, skin rash and lymphadenopathy almost immediately after re-exposure. [10] Although most patients improve following discontinuation of the medication, symptoms in some patients may flare up 3 to 4 weeks after the start of the reaction, especially after rapid withdrawal of a corticosteroid. In addition, once the culprit drug is discontinued, organs initially involved may show progressive changes or organs that were previously uninvolved may manifest involvement.
Pathophysiology of DHS
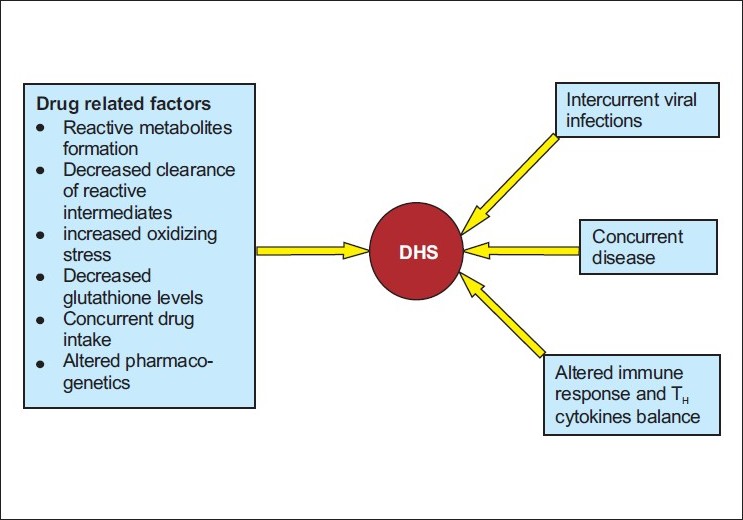
The concept that multiple factors influence the occurrence of DHS and its clinical picture and outcome has evolved over the last few centuries, especially in the areas of infectious diseases, oncology, nutritional deficiencies and autoimmune disease. [12],[13] Multiple cofactors (including a drug′s chemical and immunogenic properties along with constitutional and other environmental factors) are likely to act in concert, influencing the causation and expression of many drug reactions [Figure - 1]. Exposure to an associated drug is necessary for the occurrence of DHS; however, this exposure is not sufficient on its own. The occurrence of this syndrome is determined by the combination of a susceptible individual and exposure to a drug capable of causing this type of reaction. This is emphasized by 2 observations: (1) drugs associated with this reaction continue to be widely prescribed; (2) the frequency of idiosyncratic drug reactions (such as DHS) ranges from 1 in 100 to 1 in 10,000 people. Drug hypersensitivity syndrome usually occurs 1 to 8 weeks into the first course of therapy with the associated medication. This contrasts with other hypersensitivity reactions such as IgE-mediated anaphylaxis to penicillin, which can occur following multiple courses of drug therapy. What makes an individual unusually susceptible to develop this reaction on first exposure remains speculative, but a number of constitutional and acquired factors are probably involved. The sequence or timing of events and interaction of cofactors are also likely to be important in modifying the risk and expression of disease.
 |
| Figure 1 :Factors involved in the pathogenesis |
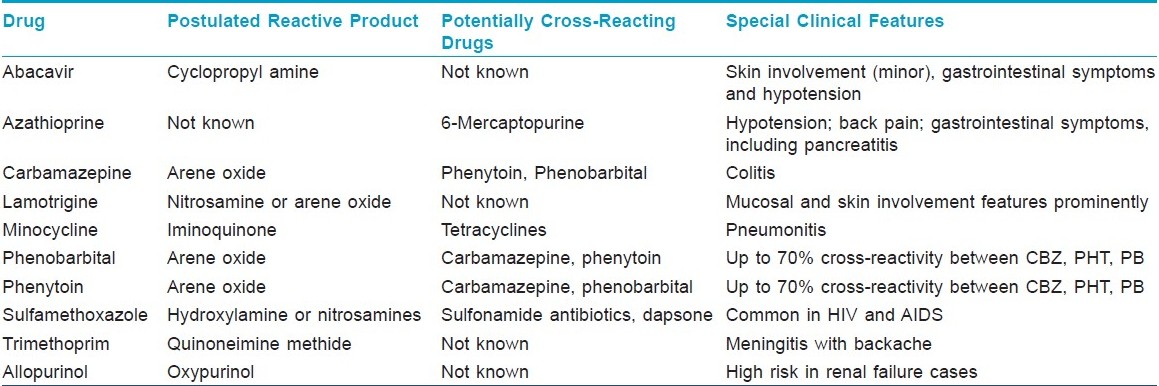
1. Reactive metabolite formation [Table - 3] and inherited genetic defects to metabolize these drugs and intermediates. Excess reactive metabolite formation has been implicated in the pathogenesis of DHS induced by PHT, CBZ or PB. These toxic metabolites can occur as a result of oxidative metabolism of the parent compounds by cytochrome P450 and other oxidative enzyme systems (e.g., myeloperoxidases, thyroid peroxidases). These toxic metabolites are usually further biotransformed and detoxified by epoxide hydroxylase, which may be lacking or mutated in persons who develop DHS. [12] However, a genetic defect altering the structure and function of epoxide hydroxylase is unlikely to be responsible for predisposing patients to AED adverse reactions. There are two hypotheses to explain this: the hapten hypothesis and the danger hypothesis. According to the hapten hypothesis, reactive drug products bind to tissue macromolecules to form complete immunogens or lead to the production of neoantigens; the danger hypothesis may explain why a potentially destructive immune response occurs instead of drug tolerance in DHS. Oxidative cell damage caused by the generation of reactive drug species could also cause or contribute to the release of cytokines that warn the immune system of cellular stress and damage. These signs of danger, also postulated to occur in a variety of conditions (including intracellular infections), promote an immune response to eliminate these modified, potentially damaged and dangerous cells. However, other theories have been proposed, e.g., that the DHS represents a form of graft-versus-host disease or that the reaction is mediated in part by circulating antibodies.
2. Intercurrent disease processes involving specific organs may increase the local toxic effects of reactive drug products and/ or facilitate production of local danger signals which influence the distribution and severity of organ involvement in DHS. Anecdotal localizing factors include prominent pneumonitis in patients with carbamazepine-associated DHS, with asymptomatic pulmonary tuberculosis or poorly controlled asthma. [13]
3. An interaction between drugs and active viral infection has been incriminated in the pathogenesis of several iatrogenic diseases, including the increased frequency of drug eruptions in individuals infected with human immunodeficiency virus (HIV). The increased occurrence of drug eruptions in several viral diseases has been attributed to stimulation and dysregulation of the immune system. The possible association of drug hypersensitivity reaction with HHV-6 and 7, cytomegalovirus and Epstein-Barr virus has been suggested by Descamps et al,[24] Changes in drug metabolism and oxidant stress may also be important factors in DHS. Rieder et al,[25] postulated that the cells from HIV-infected individuals have a higher susceptibility to the toxic effects of oxidative drug metabolites than those from controls. The addition of glutathione (a key component of the cell′s antioxidant defense) protected the lymphocytes of HIV-infected individuals against the toxic effects of these reactive drug products. Reduced levels of glutathione, selenium and other potentially important antioxidant nutrients occur in acutely ill patients with HIV.
4. There is also evidence that the cytokine profile in DHS is dynamic and may reflect changes in the T H 1/T H 2 balance over time. This suggests that changes in the T H profile may be responsible for the evolution of the disease, which is consistent with the postulated instability of early mixed T H responses. Role of IL-5 has been implicated, as the levels are raised and IL-5 is involved in development, recruitment, activation and survival of eosinophils. [12]
5. The pi- concept: [26] Drugs are not only immunogenic because of their chemical reactivity but also because they may bind in a labile way to available TCRs and possibly MHC molecules. This seems to be sufficient to stimulate certain, probably pre-activated T cells. The drug seems to bind first to the fitting TCR, which already exerts some activation. For full activation, an additional interaction of the TCR with the MHC molecules is needed. In some patients with drug hypersensitivity, within few hours of the first exposure to the drug, peptide-specific T cells are stimulated by the drug.
Diagnostic Tests
Histopathology
The most common finding on histologic examination is a dense, superficial perivascular lymphocytic infiltrate, spongiotic or lichenoid dermatitis; and, depending on the lesion biopsied, variable degrees of edema. A biopsy specimen usually shows benign lymphoid hyperplasia with preservation of normal lymph-node architecture. The lymph node biopsy specimen may show pseudolymphoma with atypical hyperplasia simulating malignancy that resolves on discontinuation of the drug. In most cases, the reticulum cell hyperplasia, loss of nodal architecture, and abnormal cells with frequent mitotic figures are not considered premalignant. In a few patients treated with phenytoin, frank lymphoma has been reported to develop.
Lymphocyte toxicity assay
The lymphocyte toxicity assay is an in vitro drug metabolite toxicity system that was developed in the early 1980s. [14] It uses murine hepatic microsomes as a source of cytochrome P450, which are incubated with the drug in question to generate reactive metabolites. Human lymphocytes are used as surrogate peripheral target cells to investigate individuals susceptible to drug toxicity. Lymphocytes are used since they are readily accessible; do not contain the enzymes which produce toxic metabolites from the parent drug, and contain detoxification enzymes (e.g. epoxide hydrolases). If the cells are susceptible to damage by reactive metabolites, this cytotoxicity can be quantified by a variety of methods. However, the lymphocyte toxicity assay is expensive and cumbersome to perform; currently, it is only being used in certain research centres. [27]
Patch testing
Several studies have evaluated the usefulness of patch testing in the diagnosis of anticonvulsant hypersensitivity syndrome. However, many of these studies have shown inconsistent results. [15] If patch testing is to be performed in patients with a history of anticonvulsant hypersensitivity syndrome, 1% and 10% CBZ or PHT in petrolatum compound is recommended for patch testing. In addition, at least 2 months should elapse from the eruption to the testing date since either false-positive reactions due to increased reactivity or false-negative reactions due to a refractory state may exist. As with any diagnostic test, diagnosis is dependent on clinical recognition and judgment. In vivo and in vitro tests (with the lymphocyte toxicity assay) showed that cross-reactivity between PHT, CBZ and PB is as high as 75%. [27],[28]
Differential Diagnosis
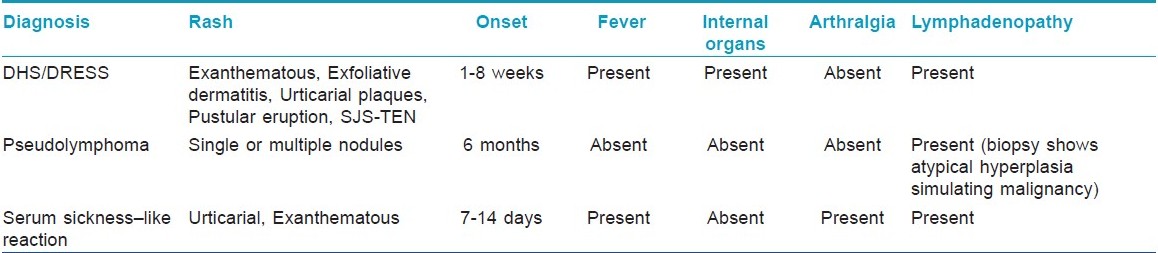
Differential diagnosis includes other cutaneous drug reactions, acute infections (e.g., Epstein-Barr virus, hepatitis virus, influenza virus, cytomegalovirus, human immunodeficiency virus, streptococcus), lymphoma or pseudolymphoma, collagen vascular diseases and serum sickness-like reaction. [15] The main distinguishing features between serum sickness-like reactions and anticonvulsant hypersensitivity syndrome are the development of arthralgias and the lack of internal organ involvement with the former [Table - 4]. Other differential diagnosis of DHS includes diseases such as Kawasaki syndrome, Still′s disease, syphilis, porphyria, hypereosinophilic syndrome; and other drug reactions to allopurinol, dapsone and sulfa drugs. Therefore, appropriate laboratory studies should be performed. Pustules in DHS differ from the typical pattern of acute generalized exanthematous pustulosis (AGEP), in which pustules are more numerous and predominant in main body folds. Exfoliative dermatitis is another clinical presentation, which may be associated with mucosal involvement, such as cheilitis, erosions, erythematous pharynx and enlarged tonsils.
Management
Discontinuation of the anticonvulsant following the development of a fever and rash, with or without lymphadenopathy, is essential to avoid potential progression of symptoms [Table - 5]. There are a minimum of laboratory tests that will help to evaluate internal organ involvement which may be asymptomatic. Liver transaminase levels, complete blood count and urinalysis and serum creatinine level should be done; in addition, the clinician should be guided by the presence of symptoms that may suggest specific internal organ involvement (e.g., respiratory symptoms). Thyroid function tests should be done, and level of antimicrosomal thyroid antibodies should be measured and repeated in 2 to 3 months. [22] A skin biopsy may be helpful if the patient has a blistering or a pustular eruption.

Antihistamines and/ or topical corticosteroids can be used to alleviate symptoms. Although the role of systemic corticosteroids is controversial, if symptoms are severe most clinicians elect to start prednisone at a dosage of 1 to 2 mg/kg/d. [29] Pulse therapy with high-dose methylprednisolone was given to a patient who developed toxic epidermal necrolysis and severe hepatitis. [30] Uncontrolled studies have shown that plasmapheresis [31] and human intravenous immunoglobulin [32] may be efficacious in the treatment of toxic epidermal necrolysis. However, these techniques have not been validated for the management of anticonvulsant hypersensitivity syndrome.
Successful carbamazepine desensitization has been carried out in 12 patients after isolated skin rash and in 1 patient with an urticarial rash and mild periorbital edema following carbamazepine administration. [33] Desensitization with oxcarbazepine was well tolerated. Desensitization is not recommended for patients with a true anticonvulsant hypersensitivity syndrome. No internal organ involvement was described in any of the patients in whom desensitization was successful.
Patients with DHS probably should not be treated with one of the arene oxide-producing anticonvulsants. Treatment with one of these anticonvulsants should not be given during the acute or convalescent period. Because the natural history of DHS may be one of remission, exacerbation or continued worsening, the use of a potentially cross-reacting anticonvulsant can confuse or worsen the clinical picture. [34],[35],[36] A benzodiazepine should be substituted for seizure control. Valproic acid is an alternative for seizure control, but because of its hepatic metabolism, it should not be used during the acute or convalescent phase. A new anticonvulsant, gabapentin, has been approved as an adjunctive therapy in the treatment of partial seizures in adults with epilepsy. It is undergoing double-blind clinical trials as a monotherapy drug. Gabapentin is not metabolized and does not contain the arene oxide structure. Therefore, it may be useful in future in patients with DHS. Lamotrigine, an antiepileptic drug of the phenyltriazine class, is chemically unrelated to existing antiepileptic drugs and is metabolized predominantly by glucuronic acid conjugation. It is indicated as adjunctive therapy in the treatment of partial seizures in adults with epilepsy.
Hematologic, hepatic and renal values should be monitored closely. Treatment consists of supportive care and close observation, with strict attention to hydration and electrolyte balance. The cutaneous manifestations respond well to topical corticosteroids, wet wraps and antihistamines. The role of systemic corticosteroids has not been studied in a randomized, placebo-controlled clinical trial. However, while corticosteroids may have a beneficial effect on the cutaneous manifestations of DHS, the internal manifestations do not seem to reverse rapidly.
Patient Counseling
After the occurrence of DHS associated with PHT, CBZ or PB, it is important to reassess the necessity for the use of an AED. If seizure control is needed, then alternative drug therapy should be chosen. It must be remembered that older aromatic AEDs exhibit high degree of cross-reactivity and that PRM is metabolized, in part, to PB. Because valproate (VPA) is dissimilar in structure to the aromatic AEDs and has been well tolerated in patients with DHS, it is usually considered as a safe alternative in these patients.
Because a hereditary component is involved in the development of DHS, first-degree relatives of patients who experienced DHS should be informed about the increased risk for DHS in response to aromatic AEDs. A positive lymphocyte toxicity assay test is very useful in clarifying an often confusing clinical situation. In vitro testing of the alternative AEDs can be helpful in guiding future therapy but may not be perfect. Relatives can be screened by using the lymphocyte toxicity assay if the original patient is positive. However, if no test is available or if results are equivocal, first-degree relatives should avoid the aromatic AEDs.
| 1. |
Bachot N, Roujeau JC. Differential diagnosis of severe cutaneous drug eruptions. Am J Clin Dermatol 2003;4:561-72.
[Google Scholar]
|
| 2. |
Sontheimer RD, Houpt KR. DIDMOHS: A proposed consensus nomenclature for the drug induced delayed multiorgan hypersensitivity syndrome. Arch Dermatol 1998;134:874-5.
[Google Scholar]
|
| 3. |
Shear NH, Spielberg SP. Anticonvulsant hypersensitivity syndrome: In vitro assessment of risk. J Clin Invest 1988;82:1826-32.
[Google Scholar]
|
| 4. |
Handfield-Jones SE, Jenkins RE, Whittaker SJ, Besse CP, McGibbon DH. The anticonvulsant hypersensitivity syndrome. Br J Dermatol 1993;129:175-7.
[Google Scholar]
|
| 5. |
Prussick R, Shear NH. Dapsone hypersensitivity syndrome. J Am Acad Dermatol 1996;35:346-9.
[Google Scholar]
|
| 6. |
Knowles SR, Shapiro L, Shear NH. Serious adverse drug reactions induced by minocycline: Report of 13 patients and review of the literature. Arch Dermatol 1996;132:934-9.
[Google Scholar]
|
| 7. |
Gupta AK, Kopstein JB, Shear NH. Hypersensitivity reaction to terbinafine. J Am Acad Dermatol 1997;36:1018-9.
[Google Scholar]
|
| 8. |
Knowles SR, Gupta AK, Shear NH, Sauder D. Azathioprine hypersensitivity-like reactions-a case report and review of the literature. Clin Exp Dermatol 1995;20:353-6.
[Google Scholar]
|
| 9. |
Arellano F, Sacristαn JA. Allopurinol hypersensitivity syndrome: A review. Ann Pharmacother 1993;19:337-43.
[Google Scholar]
|
| 10. |
Schlienger, Raymond G, Shear, Neil H. Antiepileptic drug hypersensitivity syndrome. Epilepsia 1998;39:S3-7
[Google Scholar]
|
| 11. |
Valliant L. Drug hypersensitivity syndrome: Drug rash with eosinophilia and systemic symptoms. J Dermatolog Treat 1999;10:267-72.
[Google Scholar]
|
| 12. |
Sullivan JR, Shear NH. The drug hypersensitivity syndrome: What is the pathogenesis. Arch Dermatol 2001;137:357-64.
[Google Scholar]
|
| 13. |
Licata AL, Louis ED. Anticonvulsant hypersensitivity syndrome. Compr Ther 1996;22:152-5
[Google Scholar]
|
| 14. |
Silber IB, Epstein JW. The treatment of chorea with phenylethyl-hydantoin: A study of 28 cases. Arch Pediatr 1934;51:373-82.
[Google Scholar]
|
| 15. |
Knowles SR, Shapiro L, Shear NH. Anticonvulsant hypersensitivity syndrome: Incidence prevention and management. Drug Saf 1999;21:489-501.
[Google Scholar]
|
| 16. |
Tennis P, Stern R. Risk of serious cutaneous disorders after initiation of use of phenytoin, carbamazepine, or sodium valproate: A record linkage study. Neurology 1997;49:542-6.
[Google Scholar]
|
| 17. |
Seth D, Kamat D, Monteja J. DRESS syndrome: A practical approach for primary care practitioners. Clin Paediatr (Phila) 2008;47:947-52.
[Google Scholar]
|
| 18. |
Vittorio CC, Muglia JJ. Anticonvulsant hypersensitivity syndrome. Arch Intern Med 1995;155:2285-90.
[Google Scholar]
|
| 19. |
Shear N, Spielberg S. Anticonvulsant hypersensitivity syndrome, in-vitro assessment of risk. J Clin Invest 1988;82:1826-32.
[Google Scholar]
|
| 20. |
Haruda F. Phenytoin hypersensitivity: 38 cases. Neurology 1979;29:1480-520.
[Google Scholar]
|
| 21. |
Kleier RS, Breneman DL, Boiko S. Generalized pustulation as a manifestation of the anticonvulsant hypersensitivity syndrome. Arch Dermatol 1991;127:1361-4.
[Google Scholar]
|
| 22. |
Gupta A, Eggo M, Uetrecht J, Cribb AE, Daneman D, Rieder MJ, et al. Drug induced hypothyroidism: The thyroid as a target organ in hypersensitivity to anticonvulsants and sulfonamides. Clin Pharmacol Ther 1992;51:56-7.
[Google Scholar]
|
| 23. |
Powers NG, Carson NH. Idiosyncratic reactions to phenytoin. Clin Pediatr 1987;25:120-4.
[Google Scholar]
|
| 24. |
Descamp V, Valence A, Edlinger C, Fillet AM, Grossin M, Lebrun-Vignes B, et al. Association of human herpes virus 6 infection with drug reaction with eosiniphilia and systemic symptoms. Arch Dermatol 2001;137:301-4.
[Google Scholar]
|
| 25. |
Rieder MJ, Krause R, Bird IA, Dekaban GA. Toxicity of sulfonamide-reactive metabolites in HIV-infected, HTLV-infected, and noninfected cells. J Acquir Immune Defic Syndr Hum Retrovirol 1995;8:134-40.
[Google Scholar]
|
| 26. |
Posadas SJ, Pichler WJ. Delayed drug hypersensitivity reactions - new concepts. Clin Exp Allergy 2007;37:989-99.
[Google Scholar]
|
| 27. |
Pichler WJ, Tilch J. The lymphocyte transformation test in the diagnosis of drug hypersensitivity. Allergy 2004;59:809-20.
[Google Scholar]
|
| 28. |
Mansur AT, Pekcan YS, Goktay F. Anticonvulsant hypersensitivity syndrome: Clinical and laboratory features. Int J Dermatol 2008;47:1184-9.
[Google Scholar]
|
| 29. |
Chopra S, Levell NJ, Cowley G, Gilkes JJ. Systemic corticosteroids in the phenytoin hypersensitivity syndrome. Br J Dermatol 1996;134:1109-12.
[Google Scholar]
|
| 30. |
Sheretz E, Jegasothy B, Lazarus G. Phenytoin hypersensitivity reaction presenting with toxic epidermal necrolysis and severe hepatitis. J Am Acad Dermatol 1985;12:178-81.
[Google Scholar]
|
| 31. |
Chaidemenos G, Chrysomallis F, Sombolos K, Mourellou O, Ionnides D, Papakonstantinou M. Plasmapheresis in toxic epidermal necrolysis. Int J Dermatol 1997;36:218-21.
[Google Scholar]
|
| 32. |
Viard I, Wehrli P, Bullani R, Schneider P, Holler N, Salomon D, et al. Inhibition of toxic epidermal necrolysis by blockade of CD95 with human intravenous immunoglobulin. Science 1998;282:490-3.
[Google Scholar]
|
| 33. |
Eames P. Adverse reactions to carbamazepine managed by desensitisation. Lancet 1989;1:509-10.
[Google Scholar]
|
| 34. |
Kaur S, Sarkar R, Thami G, Kanwar AJ. Anticonvulsant hypersensitivity syndrome. Paediatr Dermatol 2002;19:142-5.
[Google Scholar]
|
| 35. |
Sierra NM, Garcia B, Marco J, Plaza S, Hidalgo F, Bermejo T. Cross hypersensitivity syndrome between phenytoin and carbamazepine. Pharm World Sci 2005;27:170-4.
[Google Scholar]
|
| 36. |
Seitz CS, Pfeuffer P, Raith P, Brφcker EB, Trautmann A. Anticonvulsant hypersensitivity syndrome: Cross-reactivity with tricyclic antidepressant agents. Ann Allergy Asthma Immunol 2006;97:698-702.
[Google Scholar]
|
Fulltext Views
17,468
PDF downloads
5,645





![[Table - 1]](#tbl_ijdvl_2011_77_1_7_74964_t1.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2011_77_1_7_74964_t2.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2011_77_1_7_74964_f6.jpg){kind=link}
![[Table - 3]](#tbl_ijdvl_2011_77_1_7_74964_t3.jpg){kind=link}
![[Table - 4]](#tbl_ijdvl_2011_77_1_7_74964_t4.jpg){kind=link}
![[Table - 5]](#tbl_ijdvl_2011_77_1_7_74964_t5.jpg){kind=link}