Translate this page into:
Waardenburg syndrome: A report of three cases
Correspondence Address:
Sudip Kumar Ghosh
Department of Dermatology, Venereology, & Leprosy, R. G. Kar Medical College, 1, Khudiram Bose Sarani, Kolkata-700 004
India
| How to cite this article: Ghosh SK, Bandyopadhyay D, Ghosh A, Biswas SK, Mandal RK. Waardenburg syndrome: A report of three cases. Indian J Dermatol Venereol Leprol 2010;76:550-552 |
Abstract
Waardenburg syndrome (WS) is a rare autosomally inherited and genetically heterogeneous disorder of neural crest cell development with distinct cutaneous manifestations. Based on the clinical presentations, four subtypes of the disease are recognized. A careful clinical evaluation is required to differentiate various types of WS and other associated auditory-pigmentary syndromes. We describe a case series of WS to highlight the wide spectrum of manifestations of the syndrome including a rare association.Introduction
Waardenburg syndrome (WS) is an uncommon autosomally inherited and genetically heterogeneous disorder of neural crest cell development. WS is named after a Dutch ophthalmologist, P. J. Waardenburg, who described a syndrome comprising of six distinctive features: lateral displacement of the medial canthi and lacrimal punctae, broad and high nasal root, hypertrichosis of medial part of the eyebrows, partial or total heterochromia iridis, white forelock, and congenital deaf mutism. [1] Based on the clinical presentations, four subtypes were subsequently described. [2] WS shows no racial, ethnic, or gender predilection. According to the diagnostic criteria proposed by the Waardenburg consortium, [3] a person must have two major or one major plus two minor criteria to be diagnosed as WS type1 (WS1, MIM193500). The major criteria are sensorineural hearing loss, iris pigmentary abnormality (two eyes different color or iris bicolor or characteristic brilliant blue iris), hair hypopigmentation (white forelock or white hairs at other sites on the body), dystopia canthorum (lateral displacement of inner canthi) and the presence of a first-degree relative previously diagnosed with WS. The minor criteria are skin hypopigmentation (congenital leukoderma/white skin patches), medial eyebrow flare (synophrys), broad nasal root, hypoplasia of alae nasi, and premature graying of hairs (before age 30). However, the patients rarely display all the clinical signs. WS type 2 (WS2; MIM 193510) lacks dystopia canthorum of WS1. [2] Apart from the associated upper limb anomalies (e.g. hypoplasia, syndactyly) WS type 3 (WS3; Klein- Waardenburg syndrome, MIM 148820) is similar to WS1. [2],[4] Type 3 is the rarest form of WS. Only a few cases of WS3 have been reported since it was first described. [4] In addition to the features of WS1, type 4 WS (WS4; Shah- Waardenburg syndrome, MIM 277580) is associated with features of Hirschsprung disease. [5] We report here a case series of WS from India, for its rarity, relative paucity of reports in dermatological literature, and to emphasize a rare association of the disease.
Case Report
Our first patient was a 7-year-old Muslim boy, who had presented with fused middle and ring fingers of both hands, fused 2 nd and 3 rd toes of both the feet, and a white patch on his leg present since birth. He was born out of non-consanguineous parentage by normal vaginal delivery at full term after an uneventful pregnancy. Physical examination showed syndactyly, symmetrically involving middle and ring fingers of his hands, and 2 nd and 3 rd toes of feet. A sharply marginated, depigmented macule (8 cm Χ 7 cm) was also noted on his right shin [Figure - 1]. There was no surface change and Wood′s lamp examination showed total depigmentation of the affected area. In addition, he had dystopia canthorum, broad nasal root, synophrys, hypoplastic nasal alae, and a few white hairs on his scalp. No clinical abnormality was seen in his father. Examination of his mother (the second patient), a 38-year-old woman, revealed heterochromia iridis, dystopia canthorum, broad nasal root, hypoplastic nasal alae, synophrys, [Figure - 2] white forelock, [Figure - 3] two vitiligo-like macules on trunk, and sensorineural deafness. Detailed family history revealed that many of the family members had heterochromia iridis, premature canitis, white forelock, and sensory neural deafness in various combinations [Figure - 4].
 |
| Figure 1 :(a) Syndactyly of middle and ring fingers of the hands and the 2nd and 3rd toes of feet along with a sharply marginated, depigmented macule on the right shin. (b) Syndactyly of 2nd and 3rd toes of the left feet. (c) Syndactyly of 2nd and 3rd toes of the right feet. (d) Close-up showing fused 2nd and 3rd toes of the right feet |
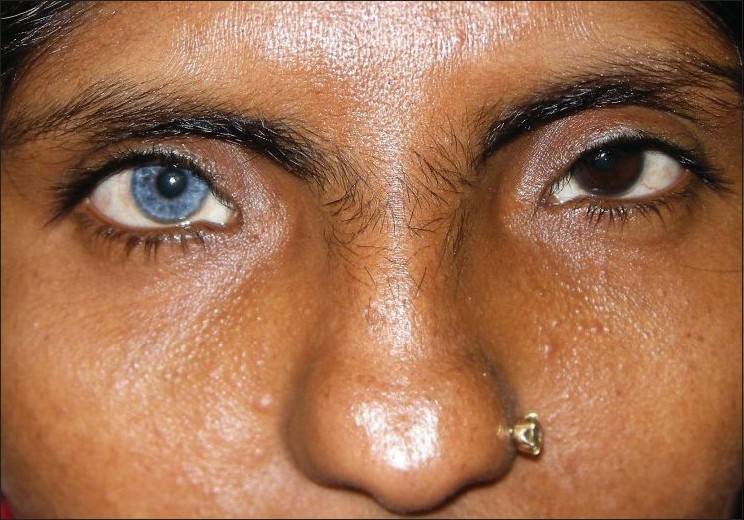 |
| Figure 2 :Showing heterochromia iridis, dystopia canthorum, broad nasal root, hypoplastic nasal alae, and synophrys |
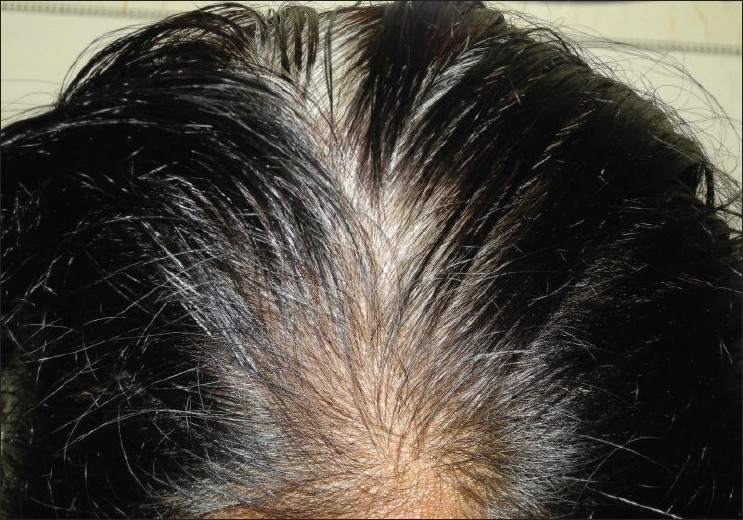 |
| Figure 3 :White forelock on frontal area |
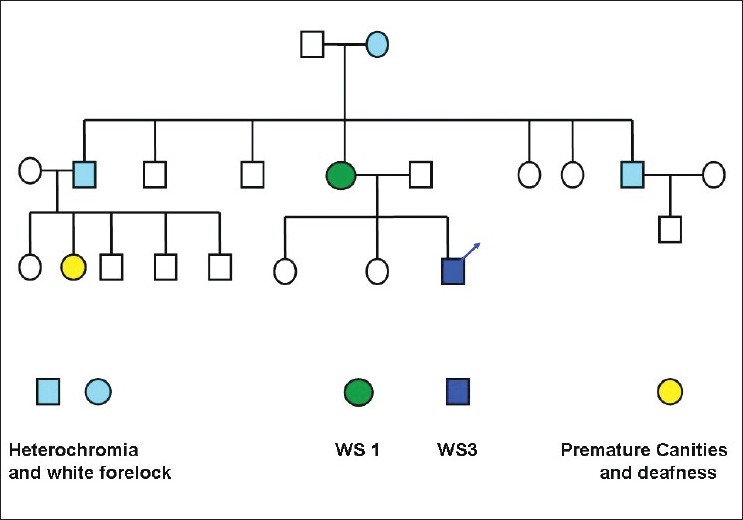 |
| Figure 4 :Showing the pedigree chart of the first patient |
The third patient was an 11-year-old Hindu boy who was brought by his parents to the dermatology clinic with the complaints of a white patch and premature graying of hairs. Examination showed a depigmented macule (6 cm Χ 6 cm) on his forearm, white forelock, heterochromia iridis, dystopia canthorum, synophrys, and sensory neural deafness.
None of our patients had mental abnormality or gastrointestinal manifestation. Systemic examination and routine laboratory work-up was normal in all the patients. Based on the clinical features the first patient was diagnosed as having WS3 as this patient fulfilled the criteria of WS1 in addition to having upper limb anomaly (syndactyly). The second and the third patient had features of WS1.
Discussion
WS 1 and 2 are autosomal dominant inherited in most cases, WS 3 may be sporadic or autosomal dominant, and WS4 is autosomal recessive in inheritance. Mutations in PAX3 gene on chromosome 2q37 are seen in WS 1 and WS 3 [2],[6] and MITF mapped on 3p12-p 14.1 are mutated in type 2 WS. [6] WS type 4 is due to SOX10 or endothelin-B receptor (EDNRB) gene mutations. [7] Apart from syndactyly, other upper limb anomalies described with WS3 include hypoplasia of the musculoskeletal system, flexion contractures, fusion of the carpal bones, and winged scapulae. [8] Most patients with WS3 have had milder anomalies of the upper extremity. [4] However, the presence of syndactyly of the feet, as seen in our first patient, has very rarely been reported in case of WS. [9] Ophthalmo-acromelic syndrome (OAS) [10] may be a close differential diagnosis of our first patient. However, in contrast to the findings of our patient, small palpebral fissure, anophthalmia, [10] flared up nostrils, bony abnormalities like the presence of only four toes [10] bilaterally and wide gap between 1st and 2nd toes are usually seen in OAS. Moreover, in OAS, eyebrows and eyelashes remain normal and depigmentation of skin and hair are not seen.
The mother of the first patient had all the classical features fulfilling the diagnostic criteria of WS1. The first two cases also demonstrated that the same family could harbor the features of both WS1 and WS3. The presence of different manifestations of WS in different combinations in the other family members of the first patient represented the well-known variable expressivity of the disease.
The third patient of this series had fulfilled the diagnostic criteria of WS1. However, in this case, history of similar illness or findings suggesting WS was not present in the other family members. Different features of WS 1 in this patient probably resulted from a new mutation. The diagnosis of WS is essentially clinical. Not every case expresses all clinical manifestations of the complete WS, and forme fruste conditions are also relatively common. [11] In individuals with only subtle features of the disease, WS may remain undiagnosed until another family member with features of WS seeks medical attention. Our case series also emphasizes the importance of examination of all the family members to identify undiagnosed cases. There is currently no definitive treatment or cure for WS. Sensorineural deafness, bony abnormalities, and Hirschsprung disease associated with WS are some of the potentially serious conditions and significantly deteriorate the quality of life. From a dermatologist′s point of view, an early diagnosis of WS from the cutaneous features may aid in the initiation of early treatment, social and vocational training, and rehabilitation of these patients.
| 1. |
Waardenburg PJ. A new syndrome combining developmental anomalies of the eyelids, eyebrows and nose root with pigmentary defects of the iris and head hair and with congenital deafness. Am J Hum Genet 1951;3:195-253
[Google Scholar]
|
| 2. |
Krishtul A, Galadari I. Waardenburg syndrome: Case report. Int J Dermatol 2003;42:651-2
[Google Scholar]
|
| 3. |
Farrer LA, Grundfast KM, Amos J, Arnos KS, Asher JH Jr, Beighton P, et al. Waardenburg syndrome (WS) type I is caused by defects at multiple loci, one of which is near ALPP on chromosome 2: First report of the WS consortium. Am J Hum Genet 1992;50:902-13
[Google Scholar]
|
| 4. |
Tekin M, Bodurtha JN, Nance WE, Pandya A. Waardenburg syndrome type 3 (Klein-Waardenburg syndrome) segregating with a heterozygous deletion in the paired box domain of PAX3: a simple variant or a true syndrome? Clin Genet 2001;60:301-4
[Google Scholar]
|
| 5. |
Shah SR. Waardenburg syndrome. Dermatol Online J 2008;14:19
[Google Scholar]
|
| 6. |
Tagra S, Talwar AK, Walia RS, Sidhu P. Waardenburg syndrome. Indian J Dermatol Venereol Leprol 2006;72:326
[Google Scholar]
|
| 7. |
Bondurand N, Pingault V, Goerich DE, Lemort N, Sock E, Le Caignec C, et al. Interaction among SOX10, PAX3 and MITF, three genes altered in Waardenburg syndrome. Hum Mol Genet 2000;9:1907-17
[Google Scholar]
|
| 8. |
Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 148820. Available from: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=148820 . [last accessed on 2010 Jan 20].
[Google Scholar]
|
| 9. |
Sener RN. Cranial MR imaging findings in Waardenburg syndrome: anophthalmia, and hypothalamic hamartoma. Comput Med Imaging Graph 1998;22:409-11
[Google Scholar]
|
| 10. |
Caksen H, Odabas D, Oner AF, Abuhandan M, Calebi V. Ophthalmo-acromelic syndrome in a Turkish infant: case report. East Afr Med J 2002;79:339-40
[Google Scholar]
|
| 11. |
Dourmishev AL, Dourmishev LA, Schwartz RA, Janniger CK. Waardenburg syndrome. Int J Dermatol 1999;38:656-63
[Google Scholar]
|
Fulltext Views
6,481
PDF downloads
2,365





![[Figure - 1]](#fig_ijdvl_2010_76_5_550_69089_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2010_76_5_550_69089_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2010_76_5_550_69089_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2010_76_5_550_69089_f4.jpg){kind=link}