Translate this page into:
Childhood sclerodermatomyositis with generalized morphea
Correspondence Address:
Rachita S Dhurat
B 14/2 Maitri park CHS, Sion Trombay Road, Chembur, Mumbai - 400 071
India
| How to cite this article: Ambade GR, Dhurat RS, Lade N, Jerajani HR. Childhood sclerodermatomyositis with generalized morphea. Indian J Dermatol Venereol Leprol 2008;74:148-150 |
Abstract
Systemic sclerosis (SS) and dermatomyositis (DM) are both multisystem disorders and share some common clinical features. We report here an 11 year-old girl whose disease showed a changing clinical pattern from juvenile systemic sclerosis (JSS) to slowly progressing juvenile dermatomyositis (JDM) and had associated generalized morphea. Serological studies revealed antinuclear antibodies (ANA) with a speckled pattern. Topoisomerase-I (Scl-70), U1 RNP (ribonucleoprotein), anti-Ro, anti-La and anti Jo-1 antibody tests were negative. Electromyography (EMG) was suggestive of primary muscle disease and histopathological findings indicated scleroderma. The patient fulfilled the American College Rheumatology (ACR) diagnostic criteria for JSS as well as Bohan and Peter criteria for JDM separately and hence, was diagnosed to have sclerodermatomyositis (SDM). Mixed connective tissue disease (MCTD) and antisynthetase antibody syndrome (ASS) which share same clinical features with SS and DM were excluded by immunological studies.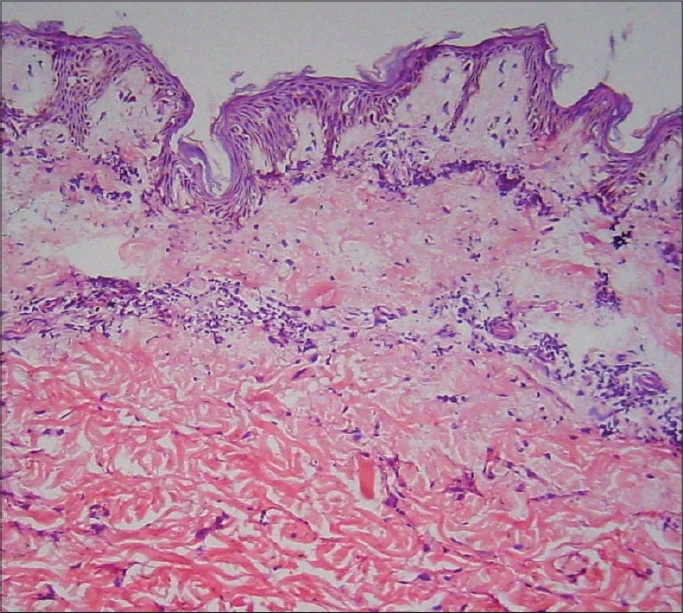 |
| Figure 3: Normal epidermis, upper dermal edema and hyalinization of collagen bundles in mid-reticular dermis (H and E stain, �100) |
|
| Figure 3: Normal epidermis, upper dermal edema and hyalinization of collagen bundles in mid-reticular dermis (H and E stain, �100) |
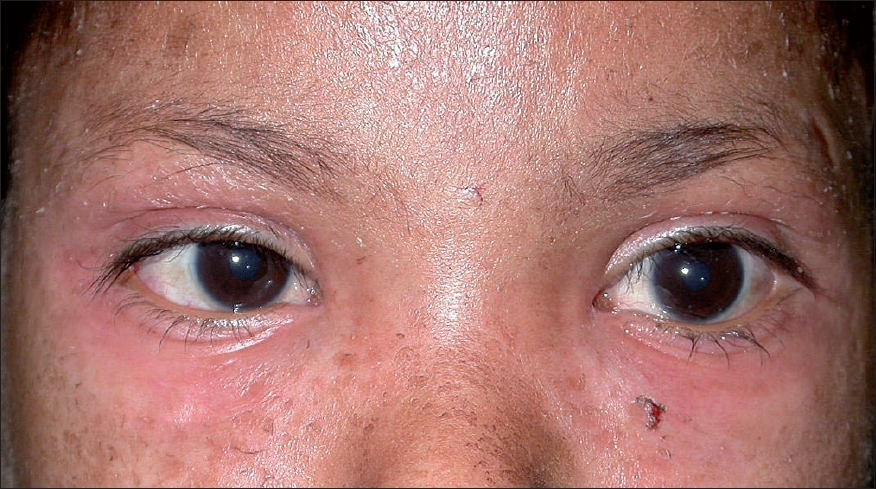 |
| Figure 2: Periorbital erythema and edema suggestive of heliotrope rash |
|
| Figure 2: Periorbital erythema and edema suggestive of heliotrope rash |
 |
| Figure 1: Bilaterally symmetrical erythematous, scaly atrophic plaques of morphea on the back |
|
| Figure 1: Bilaterally symmetrical erythematous, scaly atrophic plaques of morphea on the back |
Introduction
The term overlap syndrome (OS) may be used when patients exhibit clinical features of two or more classical connective tissue diseases (CTDs). Such patients may not only meet diagnostic criteria for one disease but also have atypical manifestation or findings suggestive of a second CTD. These include mixed connective tissue disease (MCTD), sclerodermatomyositis (SDM) and antisynthetase antibody syndrome (ASS). Systemic sclerosis combined with DM is the most frequently seen overlap syndrome and is called sclerodermatomyositis or scleromyositis. It has been mainly described in adults with a few cases of childhood SDM. Blaszczyk et al. [1] and Vandergheynst et al. [2] have reported a case series of 14 and five children respectively with positive PM-Scl antibody. The unusual feature in our case, which was not reported in previous studies, was the presence of multiple plaques of morphea.
Case Report
An 11 year-old girl presented with multiple, red, raised, scaly lesions of variable size and of two years′ duration on the chest, abdomen, back, buttocks and the extremities. At the age of ten years, she developed tightening and thickening of the skin of acral areas, which gradually progressed to involve the forearms, trunk and face. There was difficulty in opening the mouth, exertional dyspnea and intolerance to cold. A few months later, she developed moderate grade fever of sudden onset, photosensitivity, periorbital redness and swelling as well as proximal muscle weakness.
Cutaneous examination revealed multiple, bilaterally symmetrical, erythematous papules and scaly plaques with central atrophy on the chest, abdomen, buttocks and extremities [Figure - 1]. Sclerodermoid changes were present on the distal limbs with further involvement of the forearms, trunk and face. She had microstomy, flexion contracture at the elbows, edematous fingers and telangiectasias on the palms and a hard palate. She also had heliotrope rash [Figure - 2], photosensitivity and intolerance to cold without any visible Raynaud′s phenomenon. Muscle power was of grade IV and III in the shoulder and pelvic girdle respectively. She did not have any stellate scars, acro-osteolysis or Gottron′s papules and Gottron sign was negative.
On investigations, lactate dehydrogenase (LDH) was raised to 595 IU/L (Normal: 140-300 IU/L) while other muscle enzymes like creatine phosphokinase (CPK), alanine transferase (ALT), aspartate transferase (AST) and aldolase were within normal limits. Electromyography (EMG) revealed features suggestive of myositis. Histopathology performed for the atrophic erythematous plaque showed a normal epidermis, upper dermal edema and hyalinization of collagen bundles in midreticular dermis suggestive of scleroderma [Figure - 3].
Immunological studies revealed positive ANA (1+) with a speckled pattern. Double-stranded DNA, anti-Scl-70, anti-U1-RNP, anti-Ro, anti La, anti Jo-1 and lesional direct immunofluorescence tests were negative. A test for rheumatoid factor was also negative. Polymyositis-Scleroderma antibody (PM-Scl) test was not available. Pulmonary function tests and high-resolution computed tomography (CT) of the chest revealed no abnormality. Muscle biopsy was not performed. The diagnosis of SDM was confirmed based on clinico-immunological findings. The patient was treated with oral prednisolone (20 mg/day), methotrexate (5 mg/week), hydroxychloroquine (100 mg/day) and indomethacin with improvement in scleroderma lesions and muscle weakness observed over a period of six months. The raised levels of LDH reduced to normal levels after treatment and remained so on follow-up after 12 and 18 months.
Discussion
Sclerodermatomyositis has mainly been described in adults. Only a few cases of childhood SDM have been reported until now with lack of homogenous clinical pattern. [1],[2],[3] It usually starts insidiously with myalgia before the age of 12 years but sometimes, the onset may be sudden, with fever, severe myalgia, arthritis or Raynaud′s phenomenon. [4] Cutaneous features of SS and DM develop usually within the first year of illness and become pronounced during the progression of the disease. Alhough myalgia may be severe, muscle enzyme levels are usually normal or slightly raised as observed in our case. The myalgia and cutaneous signs of DM are usually transient and may regress spontaneously, whereas those of scleroderma persist. Other common features are calcinosis, arthritis and a transient Raynaud′s phenomenon that usually begin later in the course of the disease. As seen in our case, visceral involvement is often absent or mild. [1],[3],[5],[6] In SDM, the anti U1-RNP antibody is absent but other autoantibodies may be transiently detected. [3],[6],[7]
The changing clinical pattern of the disease may suggest the initial diagnosis of JRA, JSS, JDM, MCTD and ASS. [1] Juvenile systemic sclerosis often displays features of other CTDs manifesting a mild non-inflammatory myopathy with normal muscle enzyme levels. [3],[8] The differentiating features of SDM from JSS are the absence of acro-osteolysis, stellate scars, contractures of the fingers, thinning of lips, perioral furrowing, prominent telangiectasia, avascular areas on capillaroscopy and severe visceral involvement [1],[7] which were also absent in our case.
Childhood DM usually has an acute onset with raised levels of muscle enzymes and severe vascular involvement. [1],[7],[9]
The most common form of ASS is characterized by the presence of the anti-Jo 1 antibody, interstitial lung disease (ILD), inflammatory muscle disease and in many cases, fever, polyarthritis, Raynaud′s phenomenon and thick, cracked skin on the fingers (mechanic′s hand). [7],[10] Our case did not have ILD and the anti-Jo 1 antibody was absent.
Patients with MCTD, predominantly females, show features of systemic lupus erythematosus, systemic sclerosis (SLE), DM and PM. All these patients have high titers of the antibody against U1-RNP. Prominent clinical features include myositis, pulmonary hypertension, Raynaud′s phenomenon, esophageal hypomotility, swollen hands and sclerodactyly. Sclerodermatomyositis differs from MCTD by coexistent features of SS and DM with no components of SLE and absence of the anti-U1RNP antibody characteristic of MCTD. [4],[5],[7] None of the features of SLE was evident in our patient and the anti-U1RNP antibody was absent.
The features observed in our patient were diffuse scleroderma, generalized plaque type morphea, heliotrope rash, proximal muscle weakness, speckled ANA pattern, increased LDH level and EMG findings suggestive of myositis. Diagnosis of JSS, JDM, ASS and MCTD were excluded on the basis of clinico-immunological findings. Although the pattern of ANA was speckled rather than being homogeneously nucleolar which corresponds to positivity for the PM-Scl antibody, she fulfilled the Bohan and Peter′s diagnostic criteria [9] for JDM and ARA criteria for JSS [9] separately. Hence, we arrived at the final diagnosis of childhood SDM.
The identification of such patients has prognostic and therapeutic implications as these patients have a protracted and benign course. Although the clinical behavior of SDM is varied and benign, a strict follow-up for the possible onset of severe visceral involvement is nevertheless mandatory. Aggressive treatment is not usually required and the patients often respond to nonsteroidal anti-inflammatory drugs (NSAIDS) or to oral corticosteroids.
| 1. |
Blaszczyk M, Jablonska S, Szymanska-Jagiello W, Jarzabek-Chorzelska M, Chorzelski T, Mohamed AH. Childhood scleromyositis: An overlap syndrome associated with PM-Scl antibody. Pediatr Dermatol 1991;8:1-8.
[Google Scholar]
|
| 2. |
Vandergheynst F, Ocmant A, Sordet C, Humbel RL, Goetz J, Roufosse F, et al. Anti-pm/scl antibodies in connective tissue disease: Clinical and biological assessment of 14 patients. Clin Exp Rheumatol 2006;24:129-33.
[Google Scholar]
|
| 3. |
Oddis CV, Okano Y, Rudert WA, Trucco M, Duquesnoy RJ, Medsger TA Jr. et al. Serum antibody to the nucleolar antigen PM-Scl. Arthritis Rheum 1992;35:1211-7.
[Google Scholar]
|
| 4. |
Garcia-Patos V, Bartralot R, Fonollosa V, Arnal C, Boronat M, Gelpi C, et al. Childhood sclerodermatomyositis: Report of a case with anti PM-Scl antibody and mechanics hand. Br J Dermatol 1996;135:613-6.
[Google Scholar]
|
| 5. |
Mimori T. Scleroderma-polymyositis overlap syndrome. Int J Dermatol 1987;26:419-25.
[Google Scholar]
|
| 6. |
Genth E, Mierau R, Genetsky P, von Muhlen CA, Kaufmann S, von Wilmowsky H, et al. Immunogenic association of scleroderma related antinuclear antibody. Arthritis Rheumat 1990;33:657-65.
[Google Scholar]
|
| 7. |
Blaszczyk M, Jarzabek-Chorzelska M, Jablonska S, Chorzelski T, Kolacinska-Strasz Z, Beutner EH, et al. Autoantibodies to nuclear antigens in systemic scleroderma: Clinical correlation. Br J Dermatol 1990;123:421-30.
[Google Scholar]
|
| 8. |
Zulian F. Scleroderma in Children. Pediatr Clin North Am 2005;52:521-45.
[Google Scholar]
|
| 9. |
Kevin J. Juvenile dermatomyositis: Advances in understanding and management. APLAR (Asia Pacific League of Association of Rheumatology) J Rheumat 2003;6:50-63.
[Google Scholar]
|
| 10. |
Brian M, Compeyrot-Lacassagne S. Inflammatory myopathies in children. Pediatr Clin North Am 2005;52:493-520.
[Google Scholar]
|
Fulltext Views
3,661
PDF downloads
1,344





![[Figure - 1]](#fig_ijdvl_2008_74_2_148_39702_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2008_74_2_148_39702_2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2008_74_2_148_39702_3.jpg){kind=link}