Translate this page into:
Waardenburg syndrome type 2 in an african patient
Correspondence Address:
S GH Otman
Department of Dermatology, University Hospital of Wales, Heath Park, Cardiff, CF 14 4XN
United Kingdom
| How to cite this article: Otman S G, Abdelhamid N I. Waardenburg syndrome type 2 in an african patient. Indian J Dermatol Venereol Leprol 2005;71:426-427 |
Abstract
A thirty six year-old African man, born in the Southern part of Libya, presented with congenital deafness and white forelock, variable-sized hypopigmented, depigmented patches and hyperpigmented islands within the areas of hypomelanosis affecting the upper parts of the trunk, both arms and forearms. The nasal root was hypertrophied, but there was a lack of lateral displacement of medial canthi. We report this case of Waardenburg syndrome type 2 (WS 2). As no treatment is available for patients with WS 2, prompt diagnosis and referral to a hearing specialist are crucial for the normal development of patients affected with this condition.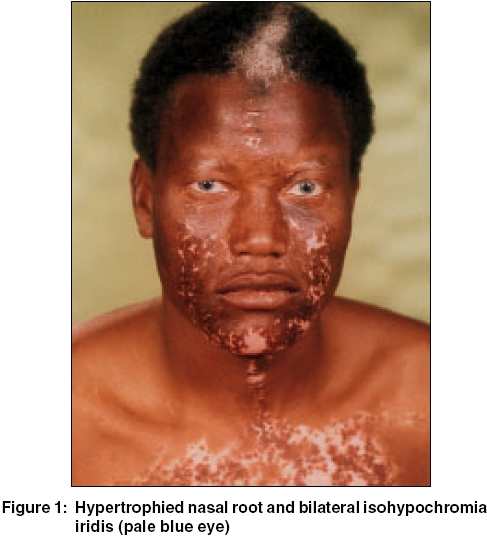 |
|
Introduction
Waardenburg syndrome (WS) is a rare, autosomal dominant disorder characterized by varying degrees of: congenital hearing loss; dystopia canthorum; synophrys; broad nasal root; depigmentation of hair (white forelock), skin or both; and heterochromic or hypochromic irides. This condition is caused by the physical absence of melanocytes in the skin, hair, eyes, and the stria vascularis of the cochlea, and is classified as a disorder of neural crest cell development. We report a case of WS type 2 in an adult who presented all the symptoms characteristic of this syndrome. A careful clinical description is useful to differentiate between various types of WS and other associated auditory-pigmentary syndromes. WS type 2, characterized by presence of white forelock, unilateral or bilateral deafness, but without the dystopia canthorum. WS 2 is a heterogeneous group, with about 15% of cases caused by mutations in microphthalmia associated transcription factor (MITF).
Case report
A thirty-six year-old African man, born in the Southern part of Libya, presented with congenital deafness and a white forelock. He was noted to have variable-sized hypopigmented and depigmented patches. There were hyperpigmented islands within the areas of hypomelanosis. The pigmentary changes extended over the upper parts of trunk, both arms, and forearms. The nasal root was hypertrophied, but there was no lateral displacement of medial canthi [Figure - 1]. Eye examination showed blue irides with iris heterochromia. Fundus examination revealed retinal hypopigmentation and pigment mottling in the periphery. Family history revealed that one of his paternal cousins was similarly affected. Consanguinity was not reported. Histologic analysis showed a decrease of melanocytes within the depigmented areas.
Discussion
Waardenburg syndrome (WS) is a hereditary auditory-pigmentary syndrome. It was described in 1947[1] by Waardenburg and Klein in 1950.[2] The major features of this syndrome are congenital sensorineural hearing loss and pigmentary disturbance of eyes, hair, and skin. The pathogenesis of the disorder is thought to be a defect in melanocyte differentiation or migration. The incidence is estimated at one in 42,000 in the Netherlands (Waardenburg 1951) and 1 in 20,000 in Kenya (Hageman 1980). It is estimated that between 1-2% of WS patients are congenitally deaf.
Waardenburg syndrome has been classified into four distinct sub-types. [4],[5],[6]WS type 1, is the classical form and is characterized by presence of white forelock, unilateral or bilateral deafness, facial deformity such as dystopia canthorum (lateral displacement of inner canthi), and a broad nasal root. Waardenburg syndrome type 1 also shows heterochromia of the iris, and confluent eyebrows. WS type 2 is characterized by the same features as WS type 1 but without the dystopia canthorum. WS type 3, is associated with upper limb deformity i.e., aplasia of the first 2 ribs, lack of differentiation of the small carpal bones, cystic formation of the sacrum, abnormalities of the arms [e.g., amyoplasia and stiffness of the joints, bilateral cutaneous syndactyly]. WS type 4, is associated with Hirschsprung′s disease.[5],[6] Six different mutations of PAX-3 gene have now been reported. Most cases of WS 1 are caused by mutations in the PAX3 gene located on the long arm of chromosome 2 (2q35). Mutations in PAX3 have also been found in patients with a WS 3 phenotype. PAX3 belongs to a family of paired-domain proteins that bind deoxyribonucleic acid (DNA) and regulate gene expression. Mutations in the microphthalmia-associated transcription factor ( MITF) gene, located on chromosome band 3p14.1-p12.3, cause some cases of WS 2.[5] Other cases of WS 2 have been linked to another locus on band 1p; still others remain unlinked to either locus.
In conclusion, we report a case of Waardenburg syndrome type 2. In WS 1 and WS 2, affected individuals have a 50% chance in each pregnancy of having an affected offspring. Clinical features can be highly variable within families and no means exists to predict whether offspring will be affected more or less severely than the parent. Photoprotection is recommended to protect the amelanotic areas from burning with sun exposure. Skin graft, minigrafts, erbium:YAG laser and grafts autologous cultured melanocytes may have a role in the management of selected cases.[6]
| 1. |
Waardenburg P. A new syndrome combining developmental abnormalities of the eye, eyebrows and nose root with pigmentary defects of the iris, head, hair and congenital deafness. Am J Hum Genet 1951;3:195-253.
[Google Scholar]
|
| 2. |
Arnvig J. The Waardenburg-Klein syndrome. Nord Med 1960;64:953-5.
[Google Scholar]
|
| 3. |
Hageman MJ. Heterogeneity of Waardenburg syndrome in Kenyan Africans. Metab Pediatr Ophthalmol 1981;4:183-4.
[Google Scholar]
|
| 4. |
Pardono E, van Bever Y, van den Ende J, Havrenne PC, Iughetti P, Maestrelli SR, et al. Waardenburg syndrome: clinical differentiation between Type I and II. Am J Med Genet 2003:117A : 223-35.
[Google Scholar]
|
| 5. |
Badner JA, Chakravarti A. Waardenburg syndrome and Hirschsprung disease: evidence for pleiotropic effect of a single dominant gene. Am J Med Genet 1990;35:100-4.
[Google Scholar]
|
| 6. |
Chang KK, Wong CK, Lui VC, Tam PK, Sham MH. Analysis of SOX10 mutations identified in Waardenburg- Hirschsprung patients: Differential effects on target gene regulation. J Cell Biochem 2003;90:573-85.
[Google Scholar]
|
Fulltext Views
4,375
PDF downloads
2,875





![[Figure - 1]](#fig_ijdvl_2005_71_6_426_18951_1.jpg){kind=link}