
Translate this page into:
A case of Conradi-Hünermann-Happle syndrome with typical clinical manifestations confirmed by genetic mutation analysis
Corresponding author: Dr. Kui Young Park, Department of Dermatology, Chung-Ang University College of Medicine, Seoul, Korea. kyky@caumc.or.kr Dr. Su Yeong Kim, Department of Pediatrics, Chung-Ang University College of Medicine, Seoul, Korea. kimsy@caumc.or.kr
-
Received: ,
Accepted: ,
How to cite this article: Hong JK, Han HS, Seo SJ, Kim SY, Park KY. A case of Conradi-Hünermann-Happle syndrome with typical clinical manifestations confirmed by genetic mutation analysis. Indian J Dermatol Venereol Leprol, doi: 10.25259/IJDVL_876_20
Sir,
Conradi-Hünermann-Happle syndrome, also known as X-linked dominant chondrodysplasia punctata (CDPX2), is a group of disorders affecting cholesterol metabolism due to mutations in the emopamil-binding protein gene encoding emopamil-binding protein, the 3-beta-hydroxysteroid-delta(8)- and delta(7)-isomerase. Patients with ConradiHünermann-Happle syndrome present with skeletal dysplasia as well as cutaneous and ocular abnormalities. The annual incidence of Conradi-Hünermann-Happle is approximately one in 400,000 births and over 95% of patients are female.
A one-day-old full-term female neonate was brought to our department with diffuse scaly patches covering almost the whole of her body. Her mother had a medical history of ichthyosis, cataracts and shortening of her left upper arm. The patient has one sister who has relatively mild clinical abnormalities including short stature and ichthyosis. On clinical examination, feather-like yellowish hyperkeratotic scales were found covering almost the entire body along the Blaschko lines [Figures 1a and 1b]. In addition, asymmetric rhizomelic shortening of the upper limbs and polydactyly with pathological short stature (<1 percentile) were also noted [Figures 1c and 1d]. Ophthalmic consultations revealed mild cortical opacities in both eyes, suggestive of early-stage cataract.
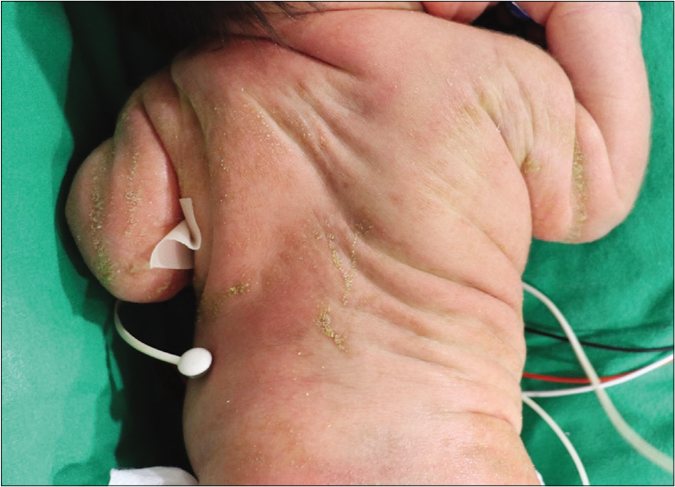
- Distinctive cutaneous manifestations with orthopedic abnormalities. Yellow-to-white hyperkeratotic scales on the back distributed along Blaschko lines
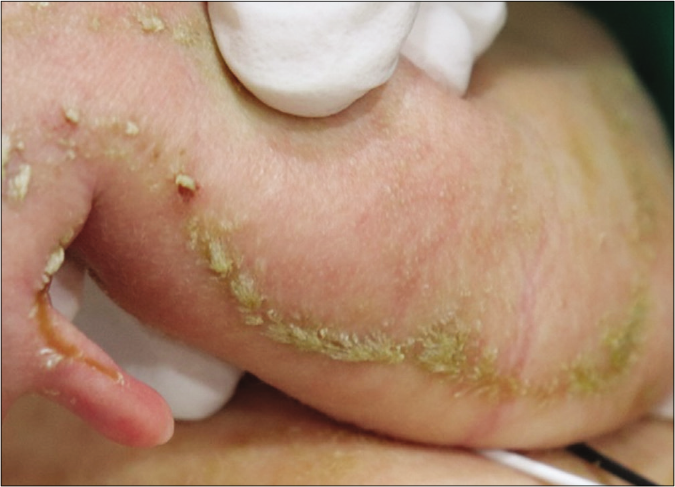
- Feather-like yellowish scales were found along the Blaschko lines
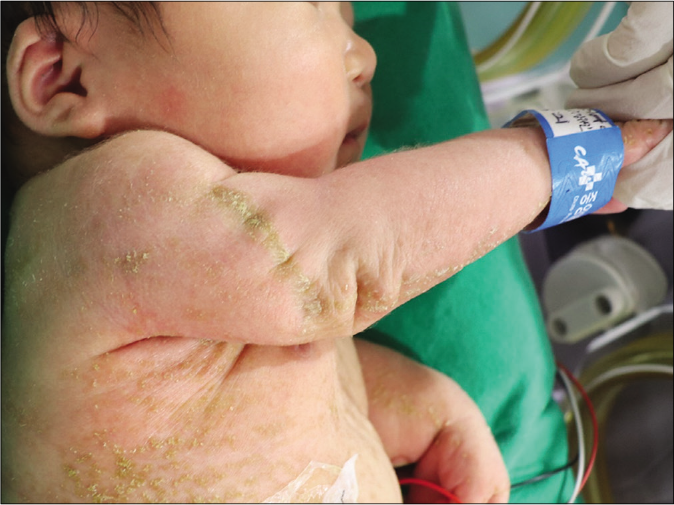
- Asymmetric rhizomelic shortening of the upper extremities (right only)
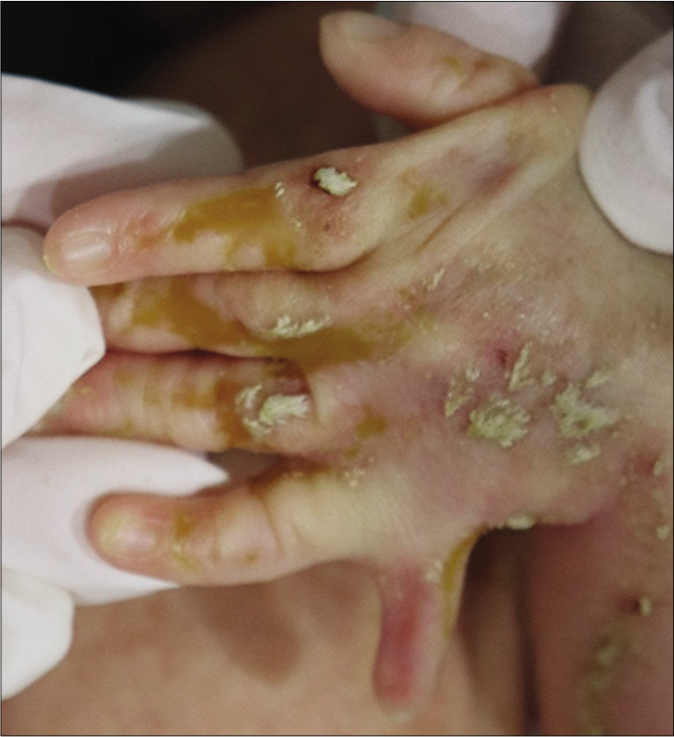
- Polydactyly of the left hand
A radiographic survey revealed punctate stippled calcification involving the multiple vertebrae, sternum, femoral epiphysis, left radius and ulna, along with scoliosis [Figures 2a-2c]. After obtaining informed consent, genomic mutation analysis was performed and it was found that the patient was heterozygous for a mutation of one guanine residue at nucleotide position 388 to thymine (c.388G>T) in the emopamil-binding protein gene with a missense mutation (p.Gly130*). This mutation is a “likely pathogenic” variant of the emopamil-binding protein gene, which causes Conradi-Hünermann-Happle syndrome.
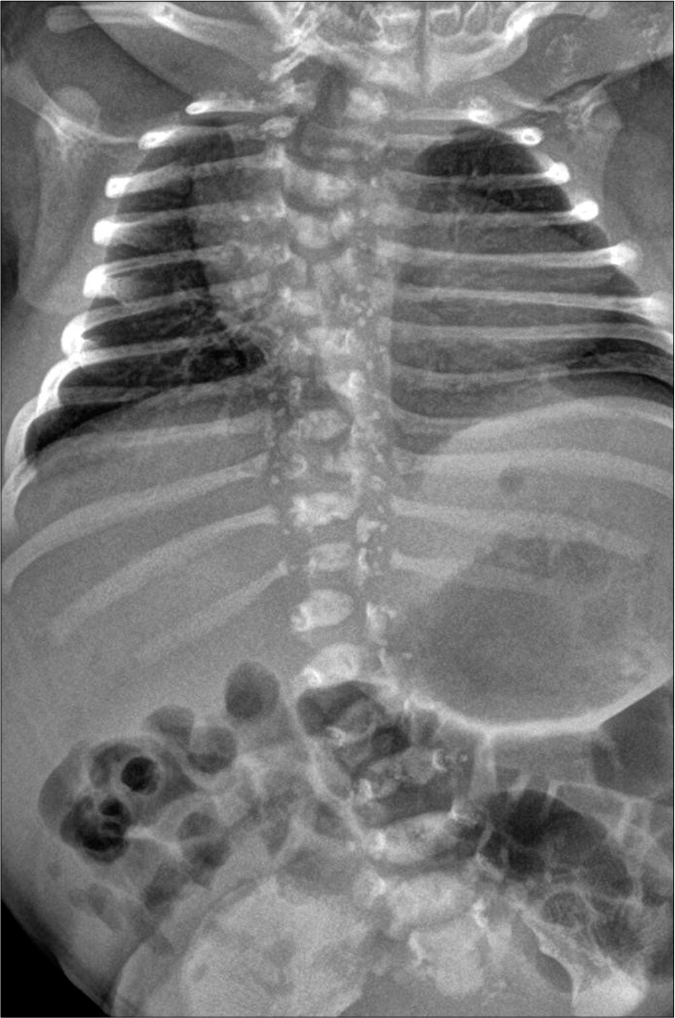
- Radiologic characteristics of the patient. Punctate stippled calcification in the vertebral bodies of multiple thoracolumbar vertebrae and sternum with scoliosis.
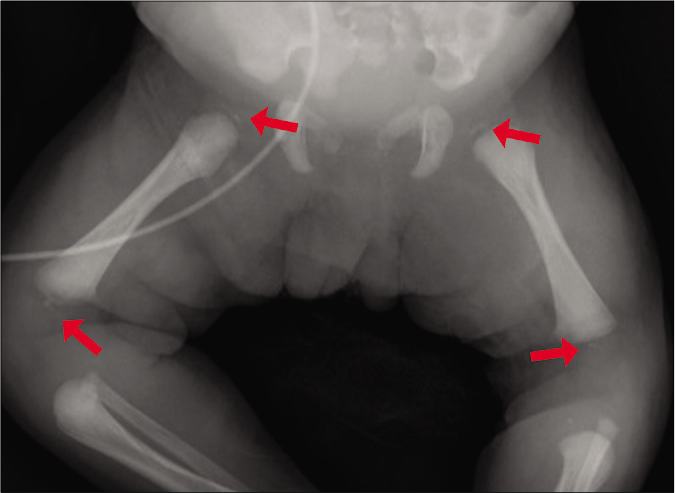
- Punctate stippled calcifications also found in both the epiphysis of the femur (red arrows)
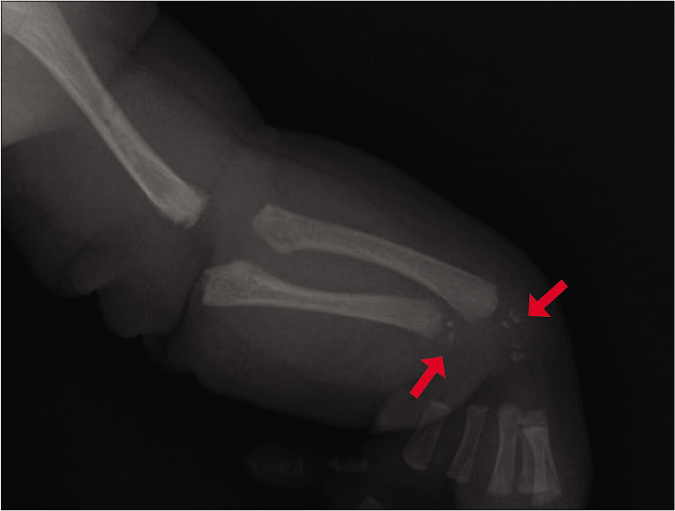
- In the radius and the ulna, multiple prominent punctate stippled calcifications were observed (red arrows)
For the treatment of ichthyosis, the parents were asked to apply moisturizers and urea cream twice a day, which cured most of the lesions, leaving linear hypopigmented lesions after about a month. Regular examinations were performed for her orthopedic and ophthalmic problems.
Chondrodysplasia punctata is a rare form of skeletal dysplasia characterized by skeletal abnormalities such as short stature, asymmetrical rhizomelic shortening of the limbs and epiphyseal stippling. Conradi-Hünermann-Happle syndrome, or CDPX2, exhibits distinctive and striking cutaneous changes including striated feather-like hyperkeratotic scales following the Blaschko lines, compared to other subtypes of chondrodysplasia punctata.
This syndrome is caused by mutations in the emopamil-binding protein gene, which encodes emopamil-binding protein that plays a key role in the final step of cholesterol biosynthesis. Enzyme deficiency leads to the accumulation of teratogenic sterol precursor compounds in tissue and serum, leading to various skeletal abnormalities.1 Furthermore, sterol and related metabolites are essential components of the cell membrane. They are also required in the hedgehog signaling pathway, which produces hedgehog protein and plays a key role in skeletal development. Therefore, dysfunction of these pathways leads to various clinical abnormalities of the skin and bony structures. To date, more than 70 emopamil-binding protein mutations have been reported, including mutations in exons and introns. These mutations induce diverse splicing pathogenic emopamil-binding protein, which could result in various phenotypic features in Conradi-Hünermann-Happle syndrome. However, the correlation between genotype and phenotype remains unclear.2,3
There are two types of congenital ichthyosis: nonsyndromic type with symptoms limited only to the skin and syndromic type with underlying genetic defects that affect other organs too. The latter includes Netherton syndrome, Sjögren-Larsson syndrome, Dorfman-Chanarin syndrome and Conradi-Hünermann-Happle syndrome. Therefore, in patients with congenital ichthyosis, other organ abnormalities should be examined to determine the type of syndrome. Furthermore, Conradi-Hünermann-Happle syndrome should be differentiated from congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome. Unlike CHILD syndrome, in Conradi-HünermannHapple syndrome, ichthyosis is bilateral and appears along Blaschko lines with a frequent co-occurrence of ocular lesions.
Interestingly, the affected family members with an abnormal X chromosome in this case showed variable phenotypic features [Figure 3]. Skewed X-inactivation is proposed as a possible mechanism of this phenomenon.4 While skewed X-inactivation can usually account for the phenotypic diversity of X-linked recessive disorders, it has also been reported to cause variable phenotypes in X-linked dominant diseases. There are a few reported cases in which skewed X-inactivation was confirmed by X-inactivation analysis in Conradi-Hünermann-Happle syndrome.4,5 However, the correlation between the severity of Conradi-HünermannHapple syndrome and the presence of skewed X-inactivation has not been established. Considering the intrafamilial variability of phenotypic findings in this case, the presence of skewed X-inactivation cannot be excluded.
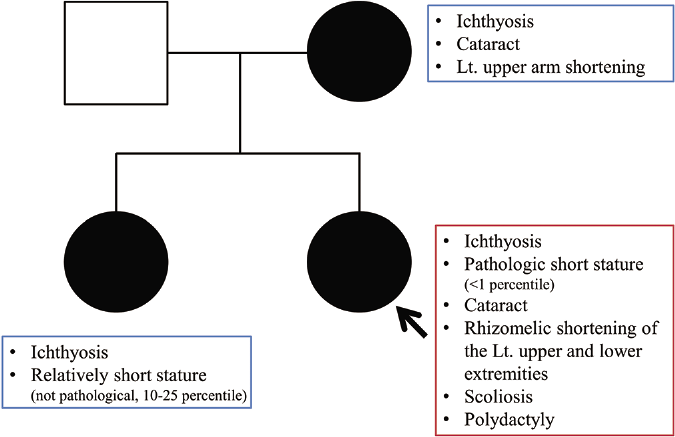
- Pedigree chart of the family showing the different phenotypic features
Mutation analysis is a powerful tool to confirm ConradiHünermann-Happle syndrome, but is often time-consuming and usually takes two to three months. Skin involvement is observed in more than 95% of cases of Conradi-Hünermann-Happle syndrome. Therefore, dermatologists should suspect ConradiHünermann-Happle syndrome on encountering patients with distinctive feathery ichthyosis and suggest additional work-up such as orthopedic and ophthalmologic evaluation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Novel EBP gene mutations in ConradiHünermann-Happle syndrome. Br J Dermatol. 2007;157:1225-9.
- [CrossRef] [Google Scholar]
- Gas chromatography-mass spectrometry and molecular genetic studies in families with the Conradi-HünermannHapple syndrome. J Invest Dermatol. 2002;118:851-8.
- [CrossRef] [Google Scholar]
- New splicing pathogenic variant in EBP causing extreme familial variability of Conradi-Hünermann-Happle syndrome. Eur J Hum Genet. 2018;26:1784-90.
- [CrossRef] [Google Scholar]
- Skewed X-chromosome inactivation causes intra-familial phenotypic variation of an EBP mutation in a family with X-linked dominant chondrodysplasia punctata. Hum Genet. 2003;112:78-83.
- [CrossRef] [Google Scholar]
- Clinical, molecular and biochemical characterization of nine Spanish families with Conradi-Hünermann-Happle syndrome: New insights into X-linked dominant chondrodysplasia punctata with a comprehensive review of the literature. Br J Dermatol. 2012;166:830-8.
- [CrossRef] [Google Scholar]




