
Translate this page into:
Severe atypical hydroa vacciniforme-like lymphoproliferative disorder in a patient with hyper IgE syndromes due to DOCK8 gene mutation
Corresponding author: Dr. Donghua Liu, Department of Dermatology and Venereology, First Affiliated Hospital of Guangxi Medical University, No. 6, Shuang Yong Road, Nanning, Guangxi, China, ldhgxmu@163.com
-
Received: ,
Accepted: ,
How to cite this article: Zhang C, Chang L, Yang X, Khan R, Liu D. Severe atypical hydroa vacciniforme-like lymphoproliferative disorder in a patient with hyper IgE syndromes due to DOCK8 gene mutation. Indian J Dermatol Venereol Leprol 2023;89:874-7.
Dear Editor,
Hyper IgE syndromes due to dedicator of cytokinesis 8 (DOCK8) gene mutations is a primary immune deficiency characterised by recurrent respiratory and skin infections, and eczema.1 Hydroa vacciniforme-like lymphoproliferative disorder is a rare skin disease caused by Epstein–Barr virus infection and the abnormal proliferation of T-lymphocytes.2 It often occurs in immunodeficient individuals. We report a teenage girl presenting with atypical hydroa vacciniforme-like lymphoproliferative disorder associated with DOCK8 mutation.
A 14-year-old girl, born of non-consanguineous marriage presented with a two-year history of erythema and blistering on the lower limbs (sun-exposed areas) following insect bites. This was followed by extensive deep ulcerations and hemorrhagic crusts with systemic symptoms that included persistent fever and weight loss. Her past medical history included eczema, allergic rhinitis, recurrent skin/respiratory tract infections (Staphylococcus aureus), allergic diarrhea and developmental delays since birth. Her parents had no similar medical history and she had no siblings. Physical examination showed height: 138 cm and weight: 25 kg. Multiple vesicles, bullae and ulcers of varying sizes were present on the trunk and extremities. Hyperpigmented and hypopigmented macules along with superficial atrophic scars were seen on the healed ulcers [Figures 1a and 1b]. She also had splenomegaly and eyelid edema. Bilateral axillary, inguinal and supraclavicular lymph nodes were palpable.
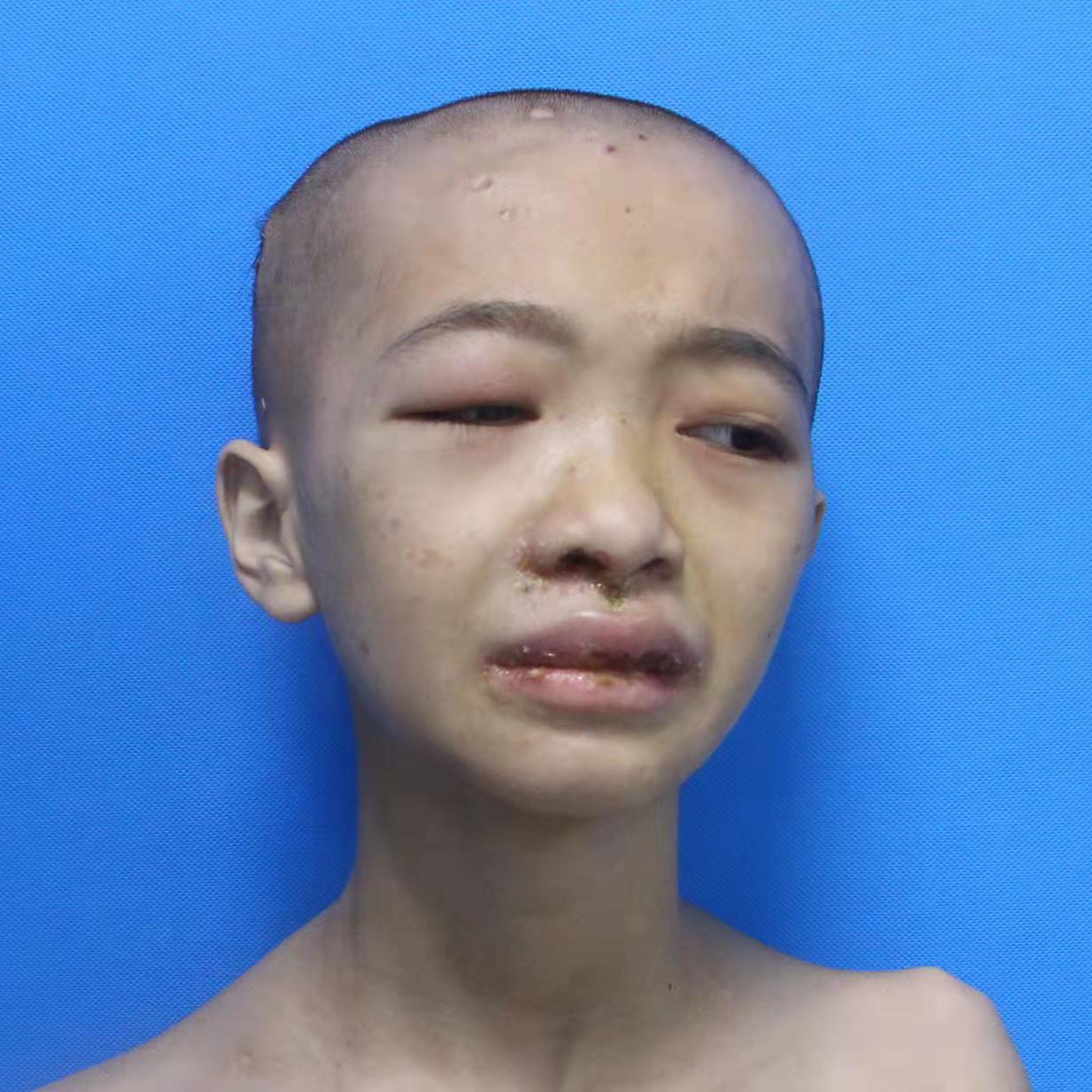
- Eyelid edema, blisters around the lip, and atrophic scars on the scalp
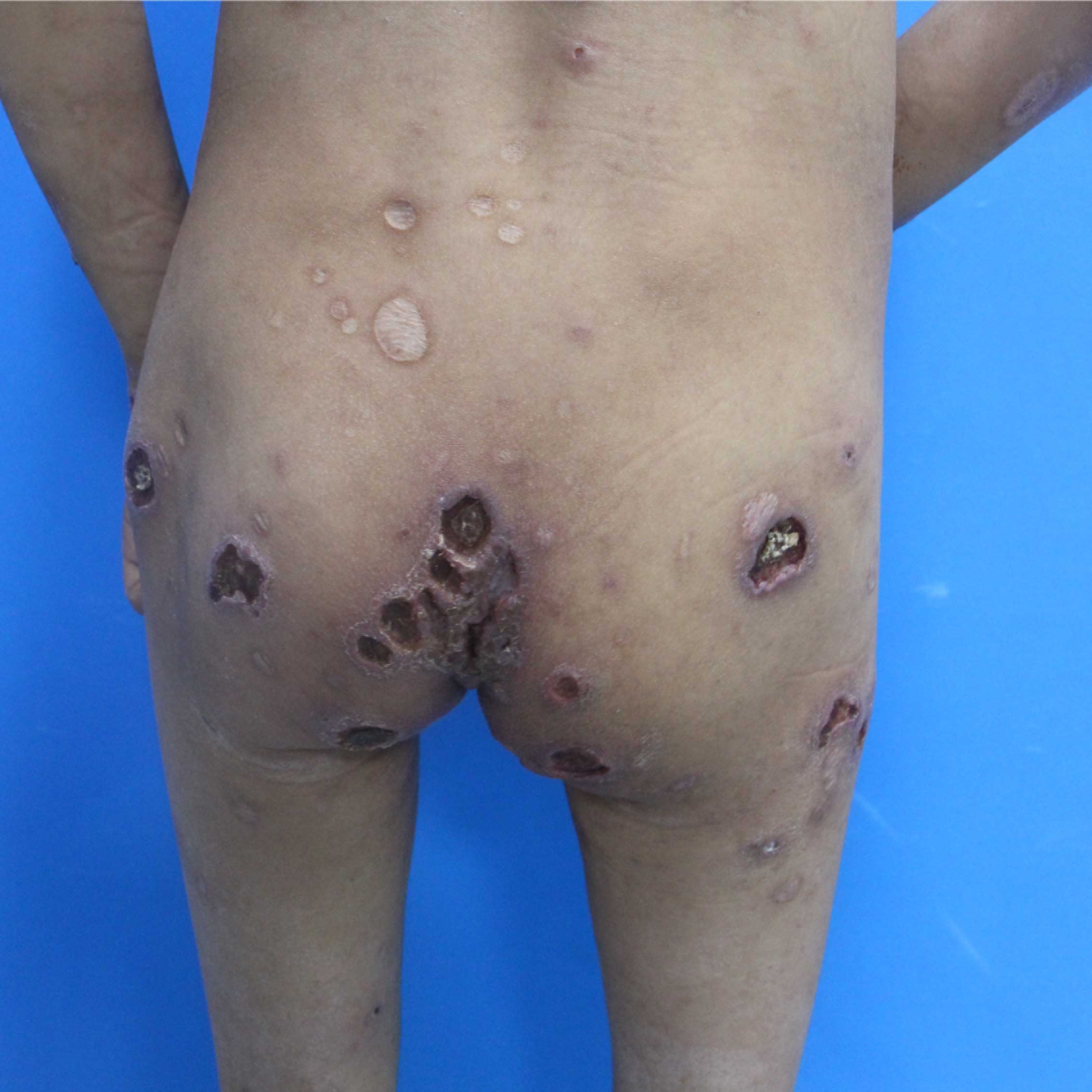
- Deep ulcers, hemorrhagic crusts, hyperpigmented or hypopigmented spots and superficial atrophic scars on the trunk and extremities
Skin biopsy from the lesion on the shin and the right inguinal lymph node showed T/natural killer cell lymphoproliferative disease [Figures 2a and 2b]. In situ hybridisation of the shin and lymph node tissue had a larger number of Epstein-Barr virus-encoded small ribonucleic acid-positive cells (EBER positive cells) [Figures 3a and 3b]. The peripheral smear was normal. Laboratory data showed a significant increase in the peripheral blood eosinophils (eosinophils 917 × 106/L; normal 50∼300 × 106/L), decreased CD 4+ T lymphocyte count (375/UL; normal 500∼1600/UL), elevated erythrocyte sedimentation rate (38 mm/hour, normal 0∼20 mm/hour) and an increase of other inflammatory indicators. The Epstein–Barr virus deoxyribonucleic acid viral titer was 4.2 × 104 IU/mL (<4.0 × 102 IU/mL) in the peripheral blood. Serum IgE titers were 5050 IU/mL (20∼200 IU/mL). T cell receptor gene rearrangement tests did not reveal monoclonal rearrangements. Phototesting or photoprovocation testing was not done. Chest computed tomography and cardiac colour ultrasound showed mild pneumonia, cardiac enlargement and pulmonary hypertension (41 mm Hg; normal 18∼25 mm Hg) suggestive of chronic respiratory infections.

- Biopsy specimen from the shin lesion shows that lymphoid cells are slightly polymorphic, with a few nuclei deeply stained and eosinophils infiltrating (hematoxylin-eosin stain, magnification ×400)
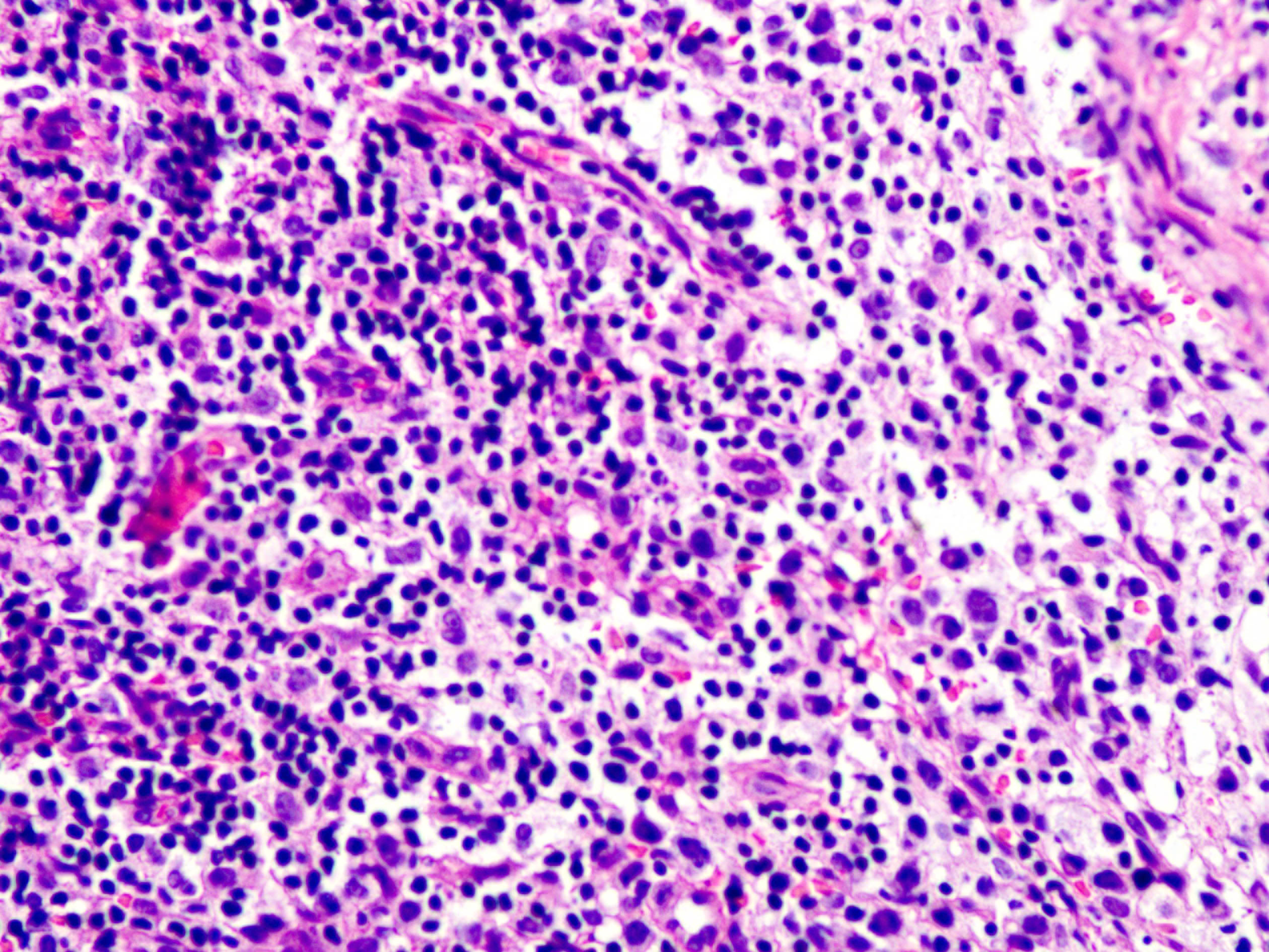
- Some lymphocyte morphologies of the right inguinal lymph node are mildly atypical, invading the follicle (hematoxylin-eosin stain, magnification ×400)

- In situ hybridisation of the shin lesion shows a large number of EBER-positive cells (magnification ×400)
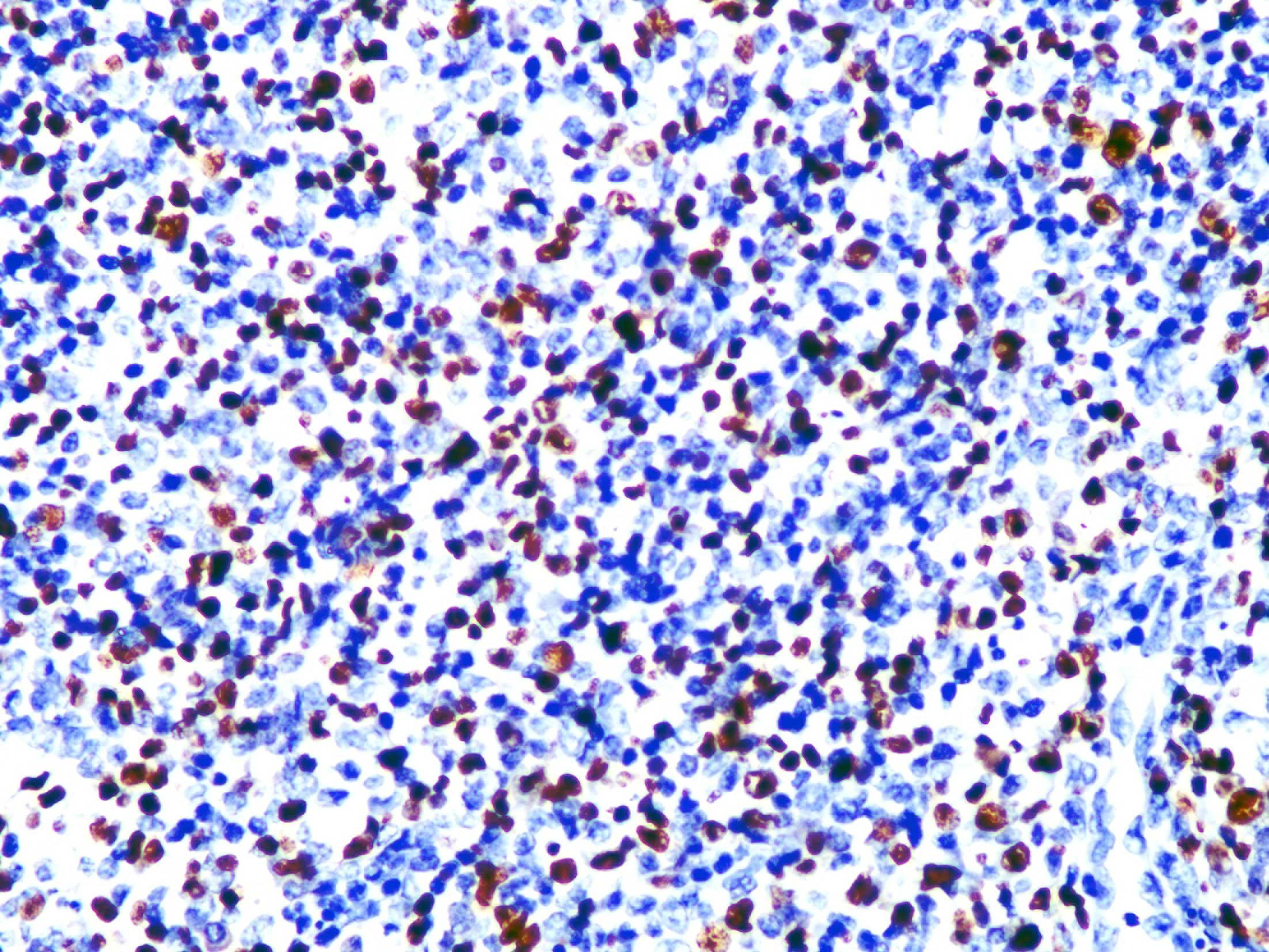
- In situ hybridisation of the tissue of the right inguinal lymph node shows EBER-positive cells (magnification ×400)
In view of the patient’s past medical history suggestive of hyper IgE syndrome, genetic sequencing of the right lymph node tissue was considered. Her family members refused genetic testing for logistic reasons. Whole-genome sequencing showed the presence of two homozygous mutations in the DOCK8 gene, two missense mutations at positions 65 and 289 in exon 3 resulting in the substitution of two amino acids of the key group (rs506121 c.65C > T, p.A22V and rs529208 c.289C > A, p.P97T). Six codon deletions in exon 39 of the lysine methyltransferase two-dimensional gene resulted in two glutamine deletions (rs765688554 c.11217_11222delGCAGCA, Gln3740_Gln 3741del).
During hospitalisation, she was treated with prednisone 15 mg/d, thalidomide 25 mg two times a day, interleukin-2500,000 U subcutaneously once a day, vancomycin 0.375 g 12 hourly and cefoperazone sodium/sulbactam sodium (2:1) 1 g given 8 hourly. Her skin condition improved during the 27 days of treatment. Subsequent visits were irregular due to COVID pandemic. Several weeks after treatment, there was recurrence of blisters, deep ulcers and Staphylococcal aureus infection on the trunk and limbs.
Mutations in the DOCK8 gene can not only lead to the development of clinical manifestations associated with hyper IgE syndromes, such as high peripheral blood IgE levels, repeated infections and developmental delay but also prevent the complete clearance of Epstein–Barr virus infection.1 Epstein–Barr virus can continue to replicate and mediate abnormal cell proliferation. DOCK8 deficiency is an important cause of Epstein–Barr virus-related lymphomas.1 Cheng and Hsu reported a case of Epstein–Barr virus-associated cutaneous natural killer/T cell lymphoma with hyper IgE syndrome whose cutaneous manifestations was similar to our case.3 However, there have been no reports of hydroa vacciniforme-like lymphoproliferative disorder associated with hyper IgE syndrome. Differential diagnosis include severe mosquito bite allergy, vasculitis, or Epstein–Barr virus-associated natural killer/T-cell cutaneous lymphomas.
Hydroa vacciniforme-like lymphoproliferative disorder includes Epstein–Barr virus-positive hydroa vacciniforme subtypes, particularly atypical or severe hydroa vacciniforme cases and hydroa vacciniforme-like lymphoma. In our case the clinical features were markedly different from those of classic hydroa vacciniforme-like lymphoproliferative disorder. Hydroa vacciniforme-like lymphoproliferative disorder mainly involves the sun-exposed areas of the skin. Typical lesions include multiple papules, plaques, blisters, ulcers, scabbing, hyperpigmented/hypopigmented spots and superficial atrophic scars.4 However, after a very short transition, our case presented with extensive lesions even on non-photo exposed sites. Eyelid edema, hepatitis, lymphadenopathy and hepatosplenomegaly were also noted. Currently, hydroa vacciniforme-like lymphoproliferative disorder is viewed on a continuum ranging from a benign and self-limited condition to a more clinically aggressive Epstein–Barr virus-associated disease.5 Therefore, a distinction between hydroa vacciniforme and hydroa vacciniforme-like lymphoma is not reliable, which means that hydroa vacciniforme-like lymphoproliferative disorder is a more appropriate diagnosis. The table from Chien-Chin Chen illustrates the differences between hydroa vacciniforme, atypical/severe hydroa vacciniforme and hydroa vacciniforme-like lymphoma [Table 1].4
Characteristics
Hydroa vacciniforme-like lymphoproliferative disorder
Classic hydroa vacciniforme
Atypical/severe hydroa vacciniforme
hydroa vacciniforme-like lymphoma
Age
Early childhood (1–7 years) and around or after puberty (12–16 years)
The median age at diagnosis is 8 years (range: 1–15 years)
Older than classic hydroa vacciniforme and atypical/severe hydroa vacciniforme
Gender
No gender bias
No gender bias
No gender bias
Race
Whites and 40–50% of nonwhites
Asians and Latin Americans
Asians and Latin Americans
Cutaneous involvement
Sun-exposed areas
Sun-exposed and sun-protected areas
Sun-exposed and sun-protected areas
Clinical presentation
Papulovesicular lesions and crusted bullae on light-exposed areas
Similar to classic hydroa vacciniforme at the early stage, but extensively recur with progression to sun-protected areas
Long-term recurrent cutaneous lesions with progression to extracutaneous involvement and haematological abnormalities
Systemic symptoms
Absent
Occasional, especially after long-term recurrence
Present (e.g., fever, hepatitis, leukopenia, lymphadenopathy, hepatosplenomegaly and hemophagocytic syndrome)
Histopathology
Reticulated epidermal degeneration and dense perivascular infiltration of mainly T cells
Similar to classic hydroa vacciniforme
Dense and extensive lymphoid infiltrate in soft tissue and extracutaneous organs. Lymphoid cells often show minimal or mild atypia
Photoprovocation
Usually positive
Often negative
Usually negative
Epstein–Barr virus deoxyribonucleic acid load (blood)
Normal
Normal or mild increased
Highly increased
Epstein-Barr virus-encoded small ribonucleic acid in situ hybridisation
Positive, mainly in T-cells
Positive, mainly in T-cells
Positive, mainly in T-cells
T cell receptor gene rearrangement
Polyclonal
Polyclonal or monoclonal
Monoclonal
Treatment
Sun protection
Sun protection, immunomodulators
Immunomodulators, haematopoetic stem cell transplantation
Prognosis
Self-limited
Chronic course with recurrent or refractory events. May evolve into hydroa vacciniforme-like lymphoma
High risk of mortality
In addition, the clinical features of this patient may also be related to the lysine methyltransferase two-dimensional gene. We found six codons missing from the patient’s lysine methyltransferase two-dimensional gene. Mutations in the lysine methyltransferase two-dimensional gene are closely associated with various lymphoproliferative diseases.6 In contrast, hydroa vacciniforme-like lymphoproliferative disorder patients without a lysine methyltransferase two-dimensional mutation showed milder symptoms with a more favourable prognosis. Genetic variation, immune deficiency and Epstein–Barr virus infection play important roles in the development of the disease, which explains why this patient’s clinical course was characterised by rapid progress and repeated recurrence.
In conclusion, severe atypical symptoms may occur in patients with hydroa vacciniforme-like lymphoproliferative disorder. The presence of genetic defects and/or immunodeficiency may be considered in this setting.
Authors Contributions
Donghua Liu proposed the conception of the work and designed the study. Li Chang and Chaoyin Zhang contributed equally to the collection of data and the writing of the manuscript. Xue Yang provided patient care. Raqib Khan performed data analysis and revised the article. Donghua Liu supervised the manuscript. All authors critically assessed the manuscript for intellectual content.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflict of interest
The authors declare no competing financial interests. The table cited in this article has been obtained with appropriate permission.
References
- DOCK8 deficiency: Insights into pathophysiology, clinical features and management. Clin Immunol. 2017;181:75-82.
- [CrossRef] [PubMed] [Google Scholar]
- Hydroa vacciniforme-like lymphoma: A chronic EBV+ lymphoproliferative disorder with risk to develop a systemic lymphoma. Blood. 2013;122:3101-10.
- [CrossRef] [PubMed] [Google Scholar]
- Hyper-IgE syndrome with Epstein-Barr virus associated extranodal NK/T cell lymphoma of skin. Kaohsiung J Med Sci. 2010;26:206-10.
- [CrossRef] [PubMed] [Google Scholar]
- Hydroa vacciniforme and hydroa vacciniforme-like lymphoproliferative disorder: A spectrum of disease phenotypes associated with ultraviolet irradiation and chronic Epstein-Barr virus Infection. Int J Mol Sci. 2020;21:9314.
- [CrossRef] [PubMed] [Google Scholar]
- Epstein-Barr virus NK and T cell lymphoproliferative disease: Report of a 2018 international meeting. Leuk Lymphoma. 2020;61:808-19.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic landscape of T- and NK-cell post-transplant lymphoproliferative disorders. Oncotarget. 2016;7:37636-48.
- [CrossRef] [PubMed] [Google Scholar]




