
Translate this page into:
A novel homozygous missense mutation in exon 3 at codon 42 c.125G>A (p.Arg42His) in the PROC gene causing protein C deficiency and presenting as neonatal purpura fulminans

Corresponding author: Dr. Aneeta Francis, Department of Dermatology & Venereology, Government Medical College, Trivandrum, India. aneetagreek@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Francis A, George AE, Ashok P, Chandran R. A novel homozygous missense mutation in exon 3 at codon 42 c.125G>A (p.Arg42His) in the PROC gene causing protein C deficiency and presenting as neonatal purpura fulminans. Indian J Dermatol Venereol Leprol. 2025;91:S79-S81. doi: 10.25259/IJDVL_618_2023
Dear Editor,
Neonatal purpura fulminans (NPF) or thrombophilia due to protein-C deficiency according to OMIM terminology, is a rapidly progressive thrombotic disorder due to congenital or acquired protein-C or protein-S deficiency characterised by haemorrhagic cutaneous necrosis and disseminated intravascular coagulation (DIC).1–3 It is a dermatological emergency and can be fatal if not managed early.3 Congenital protein-C deficiency presenting as NPF is very rare, the incidence being 1 in 4 million.1 The inheritance is autosomal recessive due to homozygous or compound heterozygous mutations in the PROC gene located on chromosome 2q13-14.2 We report a novel homozygous mutation in the PROC gene.
A term male baby (birth weight 3.1 kg), born out of third-degree consanguineous marriage to asymptomatic parents, received vitamin-K prophylaxis on the right thigh at birth and developed purpuric lesions at the injection site within 72 h. He was treated with topical heparin ointment. The lesion progressed into a necrotic eschar, but healed in 6-weeks, forming a scar. At 6 months old, a white pupil of the right eye necessitated an ophthalmic examination under anaesthesia. It revealed posterior closed funnel retinal detachment secondary to vitreous haemorrhage and persistent hyperplastic primary vitreous (PHPV) of the right eye. The baby was referred to our dermatology department for the first time, at 10 months age, with history of recurrent ecchymosis of both shins since the seventh month and painful purpuric lesions which rapidly became necrotic and indurated in a day [Figures 1, 2a, 2b and 3]. Investigations revealed anaemia [7.7 g/dl]; thrombocytopenia [77,000 cells/cu.mm]; prolonged Prothrombin Time/International Normalised Ratio (PT/INR) [64.4 seconds/5.11]; prolonged Activated Partial Thromboplastin Time (APTT) [78.5 seconds]; grossly elevated D-dimer [8.9 mcg/ml]; and reduced fibrinogen levels [131 mg/dl] consistent with disseminated intravascular coagulation (DIC). Doppler of cranial and renal vessels ruled out thrombosis. The cutaneous thrombotic manifestations along with DIC and PHPV in the absence of infectious causes or signs of septicaemia narrowed the differentials to NPF due to congenital protein-C or protein-S deficiency.
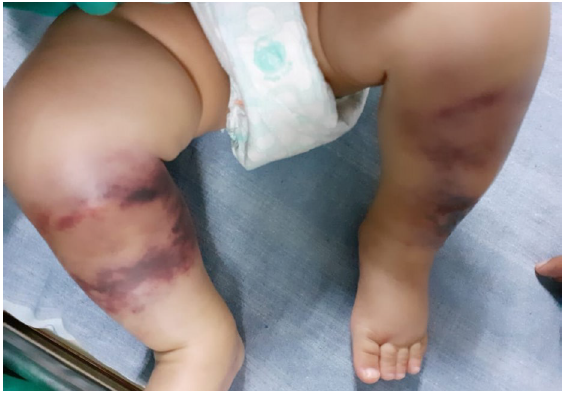
- Multiple well-defined irregular purpuric macules with areas of blackish discolouration over both legs (Day 1 of presentation – at 10 months of age).
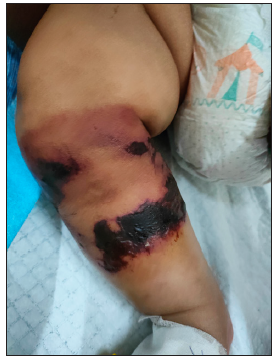
- Multiple irregular purpuric macules with areas of necrosis, blistering and blackish detachable skin over right leg (Day 2 of presentation).
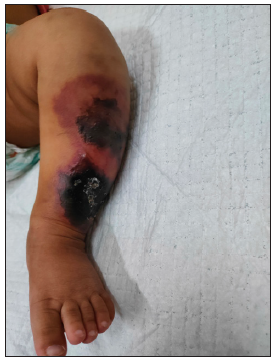
- Multiple irregular purpuric macules with central necrosis, blistering and blackish discolouration over left leg (Day 2 of presentation).
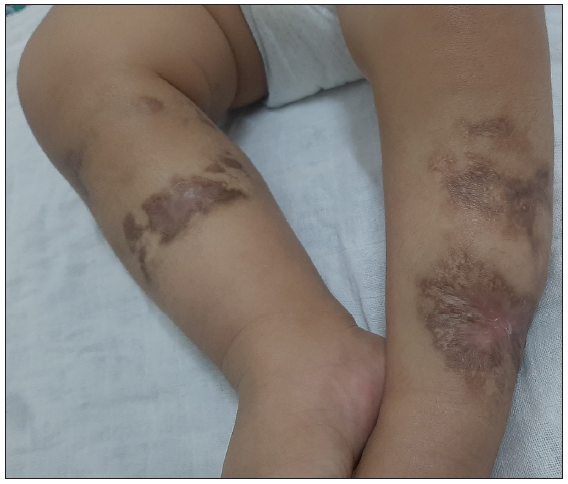
- Multiple ill-defined irregular hyperpigmented atrophic scarring with areas of depigmentation present over sites of healing. (After 4 months of maintenance therapy – 1 year and 2 months of age).
He received multiple fresh frozen plasma (FFP) transfusions for a week along with intravenous antibiotics. He was started on Inj. Enoxaparin (1 mg/kg body weight) to obtain target International Normalised Ratio (INR) of 2.5. There was a remarkable improvement in skin lesions and general condition within 3 days.
The parents denied skin biopsy. However, after obtaining informed consent, a genetic study was conducted to confirm the diagnosis by ‘massively parallel sequencing’. We detected a homozygous missense mutation in exon 3 at codon 42 c.125G>A (p.Arg42His) in the PROC gene [Table 1]. This c.125G>A variant has an allele frequency of 0.0000079% in the genome and is novel in 1000 genome databases. A final diagnosis of Neonatal Purpura Fulminans with Disseminated Intravascular Coagulation, posterior closed funnel retinal detachment secondary to vitreous haemorrhage and persistent hyperplastic primary vitreous of the right eye due to homozygous mutation in the PROC gene was made.
| Gene and Transcript | Exon/Intron number | Variant nomenclature | Zygosity | Classification | Disease | Inheritance |
|---|---|---|---|---|---|---|
| PROC (NM_000312.4) | Exon 3 | c.125G>A p. Arg42His | Homozygous | Uncertain significance | Thrombophilia due to protein C deficiency | Autosomal recessive |
The baby was continued on Inj. Enoxaparin with regular follow-up fortnightly with INR reports. No breakthrough thrombotic events occurred subsequently.
Protein-C is a vitamin-K dependant anticoagulant factor synthesised by the liver.1,2 Its activated form contains a serine protease domain and facilitates the degradation of the active forms of coagulation factors V and VIII.2
Consanguinity indicates a homozygous state which presents within the first few days of life where very low or undetectable protein-C activity (usually <1%, normal 70–140%) is seen, manifesting usually in the first few hours of birth but delayed onset between 6 and 12 months of age have also been reported.1,3,4 Heterozygous mutations in the PROC gene result in autosomal dominant thrombophilia due to protein C deficiency, usually remaining asymptomatic until adolescence or adulthood and have about 50% levels of PC.3–5 There is no clinical correlation between residual enzyme activity and clinical thrombosis as many patients with 0–1% protein C activity had NPF while one patient did not have any thrombotic episode until 11 years age.6
The cutaneous lesions are initially dark red, subsequently becoming purple-black with induration and are often mistaken as bruise.3 Limbs, buttocks and thighs are the commonest sites; affected areas may become necrotic and gangrenous and may even result in amputation.3 Systemic manifestations include thrombosis of the cerebral vasculature, vitreous haemorrhage, blindness due to retinal detachment and renal vein thrombosis.3
Management involves FFP transfusions.3,4 Highly purified protein-C concentrate (Ceprotin) is an alternative to repeated FFP transfusions.1 Patients need long-term anticoagulation therapy.3 Permanent cure may be achieved by liver transplantation from living donors.3,4 Molecular diagnosis paves way for prenatal diagnosis in subsequent pregnancies.1,2 This is pivotal, considering the poor prognosis and fatality of this dreaded condition.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- A novel mutation c.1048A>T at codon 350(Lys>Stop) in PROC gene causing neonatal purpura fulminans. Blood Coagul Fibrinolysis. 2013;24:890-2.
- [CrossRef] [PubMed] [Google Scholar]
- Management of neonatal purpura fulminans with severe protein C deficiency. Indian Pediatr. 2006;43:542-5.
- [PubMed] [Google Scholar]
- Diagnosis and management of neonatal purpura fulminans. Semin Fetal Neonatal Med. 2011;16:318-22.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis and treatment of homozygous protein C deficiency. Report of the working party on homozygous protein C deficiency of the subcommittee on protein C and protein S, international committee on thrombosis and haemostasis. J Pediatr. 1989;114:528-34.
- [CrossRef] [PubMed] [Google Scholar]
- Ophthalmic manifestations of congenital protein C deficiency: A case report and mini review. BMC Ophthalmol. 2020;20:282.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Molecular genetic analysis of severe protein C deficiency. Hum Genet. 2000;106:646-53.
- [CrossRef] [PubMed] [Google Scholar]




