
Translate this page into:
Look out for the white lesions in DOCK-8 (c. 5963 c>t) immunodeficiency – A novel mutation
Corresponding author: Dr. Neetu Bhari, Department of Dermatology and Venereology, All India Institute of Medical Sciences, New Delhi, India.
-
Received: ,
Accepted: ,
How to cite this article: Ahuja R, Sharma R, Arava S, Bhari N. Look out for the white lesions in DOCK-8 (c. 5963 c>t) immunodeficiency – A novel mutation. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_503_2024
Dear Editor,
Hyper-IgE syndrome is a rare primary immunodeficiency with both autosomal dominant (AD) and autosomal recessive (AR) inheritance. AR hyper-IgE syndrome is characterised by severe atopic eczema, a high risk for developing cutaneous viral infections and an increased propensity for malignancies.1 Herein, we describe a rare occurrence of hypopigmented lesions in a case of hyper-IgE syndrome with dedicator of cytokinesis 8 (DOCK-8) deficiency and their prognostic significance.
A 5-year-old girl, born of second-degree consanguineous marriage, presented with complaints of generalised itching for the past three years. There was a past history of frequent sinopulmonary infections including recurrent ear discharge and pneumonia requiring hospital admission. Her mother was concerned about the rapid development of relatively asymptomatic whitish lesions on the trunk for the last two months. There was also a family history of similar but fewer hypopigmented lesions in the patient’s elder brother, who had succumbed to non-Hodgkin lymphoma six months back, at the age of seven years. The mother reported a history of recurrent chest infection and empyema in him requiring multiple hospital admissions.
On examination, there was involvement of the peri-oral area, neck, cubital fossae, and popliteal fossae in the form of erythematous crusted plaques and lichenification, suggestive of atopic dermatitis. On the anterior aspect of the trunk, upper back, inguinal creases, and flexures of the extremities, barely elevated flat-topped shiny hypopigmented papules were noted [Figures 1a–1b]. These were discretely scattered on the abdomen while they tended to coalesce on the flexures. There was no lymphadenopathy, hepatosplenomegaly, or history of weight loss. Her dentition and neurological examination were unremarkable.
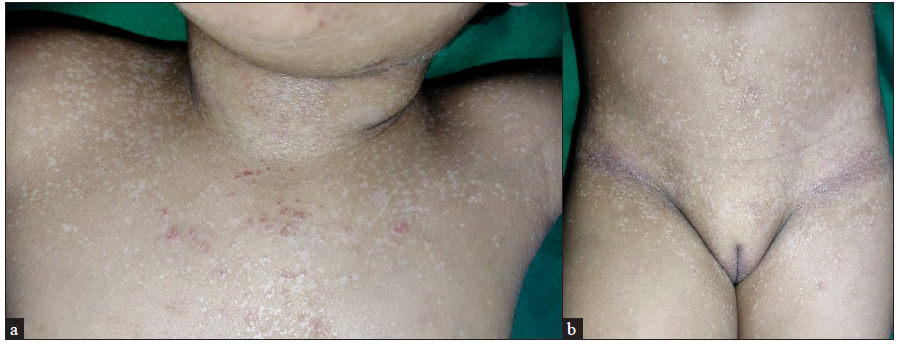
- Flat-topped shiny hypopigmented papules (a) on neck, chest and (b) on anterior trunk and lower limbs.
Investigations showed peripheral eosinophilia (13.3%; normal < 6%) with markedly raised IgE levels (99, 999 IU/mL). Skeletal survey was normal. This constellation of clinical features, history of consanguinity, family history, peripheral eosinophilia and markedly raised IgE levels prompted us to undertake genetic testing. On clinical exome sequencing, a novel pathogenic homozygous missense DOCK8 mutation c. 5963 C>T (p. Pro1898Leu) was identified. This was reported as a variant of uncertain significance as per American College of medical genetics and genomics. The in-silico predictions of this mutation were probably damaging by PolyPhen2 (HumDiv) and damaging by SIFT (sorting intolerant from tolerant), LRT (likelihood ratio test) and, MutationTaster2.
A punch biopsy from the hypopigmented papule on the back showed nests of enlarged keratinocytes in the upper epidermis with abundant bluish-grey cytoplasm, suggestive of epidermodysplasia verruciformis (EDV) [Figures 2a–2b].
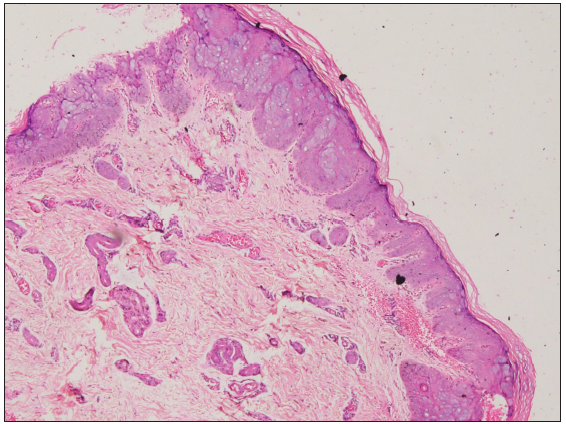
- Large, ballooned cells with bluish-grey cytoplasm in the granular and spinous layer of epidermis (Haematoxylin and eosin, 40×).
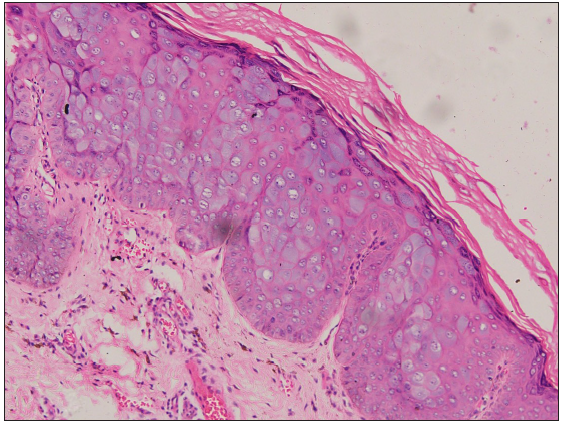
- Large, ballooned cells with bluish-grey cytoplasm in the granular and spinous layer of the epidermis (Haematoxylin and eosin, 100×).
With a diagnosis of EDV in the setting of DOCK8 deficiency, she was prescribed topical steroids, antihistamines, photoprotection, and anti-microbial prophylaxis including oral cotrimoxazole and itraconazole. She is planned for haematopoietic stem cell transplantation (HSCT) and is under follow-up in the paediatric haematology department.
Hyper IgE syndrome was traditionally classified as AD caused by a mutation in signal transducer and activation of transcription-3 (STAT-3) and AR secondary to mutations in DOCK8 and tyrosine kinase 2 gene (TYK-2).1 While parenchymal lung abnormalities, bony fractures with minimal trauma and retention of primary teeth are seen more commonly with STAT-3 deficiency, frequent upper respiratory infections, mucocutaneous candidiasis and viral infections including herpes and molluscum are present in DOCK-8 deficiency.2,3
Moreover, AR Hyper-IgE syndrome follows a more sinister course as compared to its dominant counterpart with malignancy being reported in 17% of patients at a median age of 12 years.2 These malignancies include Hodgkin and Non-Hodgkin lymphoma and squamous cell carcinomas of the skin, liver and vulva.1,3 The presence of these EDV-like lesions portends a poor prognosis with increased risk of malignancies. Till date, there are only five reports of the occurrence of EDV in AR Hyper-IgE syndrome. Like in our case, these children also had hypopigmented to erythematous flat papules on the trunk and extremities. Interestingly, two of these five children had developed cutaneous malignancies, namely, diffuse large B-cell CNS (Central Nervous System) lymphoma and metastatic multifocal leiomyosarcoma. The death of these patients occurred at the age of eight and 12 years, respectively, similar to our patient’s brother who died at seven years of age. Two other patients underwent matched donor HSCT and hence, possibly, did not report a malignant change.4,5
EDV is a pre-malignant condition with two out of every three patients developing cutaneous malignancies.6 Its occurrence in patients with DOCK8 deficiency is rarely reported and may predict the earlier development of a malignancy.
HSCT is a potentially curative treatment for patients with underlying DOCK8 deficiency; a 2-year overall survival of 84% was noted in 81 patients. Though the authors could not identify an ideal age to offer HSCT, in this cohort, the median age was 10 years.7 However, as malignancy may develop early in patients with DOCK-8 deficiency with EDV, early HSCT should be offered particularly to this patient subset.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Autosomal-recessive hyper-IgE syndrome. Indian J Dermatol. 2018;63:79-81.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The extended clinical phenotype of 64 patients with dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2015;136:402-12.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- A set of clinical and laboratory markers differentiates hyper-IgE syndrome from severe atopic dermatitis. Clin Immunol. 2021;223:108645.
- [CrossRef] [PubMed] [Google Scholar]
- A novel homozygous DOCK8 mutation associated with unusual coexistence of gross molluscum contagiosum and epidermodysplasia verruciformis in a DOCK8 deficiency patient. J Eur Acad Dermatol Venereol. 2017;31:e504-e505.
- [CrossRef] [PubMed] [Google Scholar]
- Additional diverse findings expand the clinical presentation of DOCK8 deficiency. J Clin Immunol. 2012;32:698-708.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Clinical aspects of epidermodysplasia verruciformis. J Eur Acad Dermatol Venereol. 2003;17:394.
- [CrossRef] [PubMed] [Google Scholar]
- Hematopoietic stem cell transplantation as treatment for patients with DOCK8 deficiency. J Allergy Clin Immunol Pract. 2019;7:848-55.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




