Translate this page into:
Multiplicity in clinical trial: An ignored concept
2 Department of Pharmacology, R. G. Kar Medical College, Kolkata, West Bengal, India
Correspondence Address:
Sayanta Thakur
Department of Pharmacology, Institute of Post Graduate Medical Education and Research, 244 B, A. J. C. Bose Road, Kolkata - 700 020, West Bengal
India
| How to cite this article: Thakur S, Lahiry S. Multiplicity in clinical trial: An ignored concept. Indian J Dermatol Venereol Leprol 2020;86:222-225 |
Introduction
Multiplicity is an inevitable issue in the interpretation of clinical trial data. It is defined as the potential inflation of type-I error rate (or alpha level) related to simultaneous multiple testing of different outcomes or end-points of interest.[1],[2],[3],[4]
Conventionally, most regulatory agencies accept two-sided alpha of 5% in statistical analysis. However, the alpha control strategy can vary during testing for different hypotheses, like comparing different variables at different time points between different groups. Not controlling for multiplicity can lead the investigatorto claim some of the insignificant findings as “significant” (findings will be falsely significant). Many studies with hypotheses related to secondary and exploratory objectives have used strategies to account for multiplicity.[5],[6],[7],[8] The protocol in such studies states that no final inferences will be made from these exploratory tests. Any “significant” results will be considered only “signals” of possible real effects and will have to be confirmed in subsequent studies before any final conclusions are drawn.
To control large-scale multiplicity, arising mostly in areas like genomic testing and digital image analysis, the strategy to control the false discovery rate includes avoiding even a single false conclusion of significance (as other classic alpha control methods do). In other words, one should control the proportion of tests that come out falsely positive, thereby limiting that false discovery rate to some reasonable fraction. These positive results can then be tested in a follow-up study.
Approach to a Trial With Potential Source of Multiplicity
Step 1 is to consider a wide variety of multiplicity with its source in phase III trial. Multiplicity problem can be distinguished as traditional (single-source multiplicity component) or advanced (several sources of multiplicity).[4]
Step 2 is adjusting for multiplicity, which can be related to a special situation known as a predefined or hierarchical testing sequence. It refers to sorting out end-point of interest in a sequential manner, that is, establishing a hierarchical series from the most important to the least one.[9] The testing for the most important end-point is initiated at a significance level of P < 0.05 subsequently with less important end-points until a nonsignificant result is encountered. However, if prespecification of meaningful ordering is not feasible, then the end-points of interest are tested in what is known as a data-driven testing sequence, that is, from the most significant to the least or vice versa.[9]
Information on different testing sequence could prompt the selection of the ideal test to be done for multiplicity adjustment. Specification of various tests also depends on the study design and analytical strategy. Various tests proposed for multiplicity adjustment described in the literature are illustrated in accordance with the trial scenario.[10]
Illustration of Tests for Multiplicity Adjustment in Various Trial Scenarios
There are various tests that provide the rationale for multiplicity adjustment. For instance, Bonferroni test can be justified in clinical studies where the underlying principle is to test each hypothesis at the 0.05/n alpha level.[10] A good example is a phase III study on the treatment of metastatic castration-resistant prostate cancer, where the two coprimary end-points were evaluated in terms of radiographic progression-free survival (rPFS) and overall survival (OS).[11] To control overall alpha to 0.05 across the two primary end-points, P < 0.025 was considered for significance when testing each end-point. A similar alpha splitting strategy was used in the PREVAIL trial.[12] However, the Bonferroni test has a conservative approach, and hence, newer modified approaches like Fallback test are being increasingly used. [Figure - 1] demonstrates how Fallback test could be implied upon the aforementioned scenario.[10]
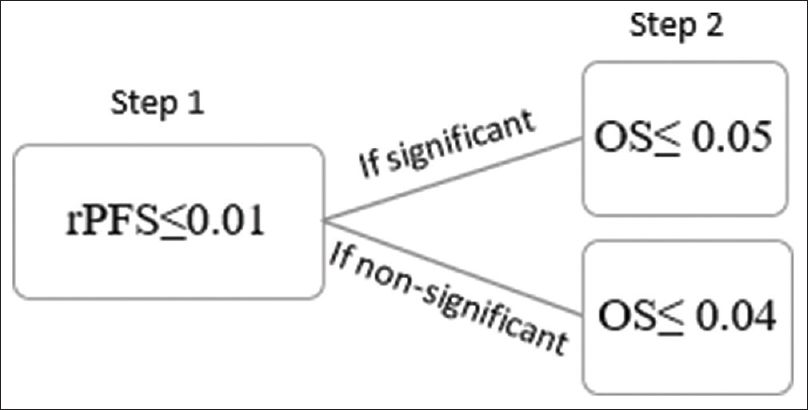 |
| Figure 1: According to Fallback's test, radiographic progression-free survival is deemed significant at Step 1 at the level of P ≤ 0.01. A treatment effect over overall survival is stated significant in Step 2 if, radiographic progression-free survival is significant in Step 1 and 0.05 for overall survival or if radiographic progression-free survival is nonsignificant in Step 1 and P ≤ 0.04 for overall survival |
In situations where there are multiple end-points to be evaluated at the same time, the Holms' test is the preferred alternative to the Fallback test. However, both the Fallback and Holms' tests are considered while evaluating three or more end-points. The testing strategy of Holms' test is represented in [Figure - 2].[10]
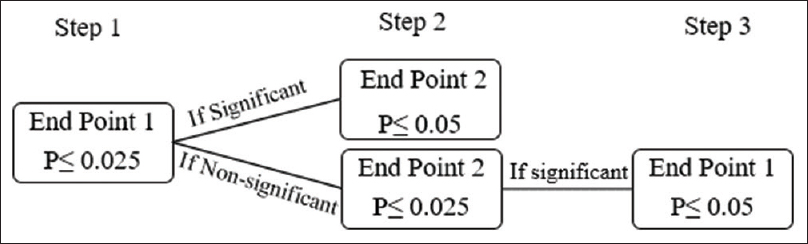 |
| Figure 2: Holms' test simulated: A significant treatment effect in Step 1 is established at P ≤ 0.025. In Step 2, end-point 2 is deemed significant if end-point 1 is significant in Step 1 and P ≤ 0.05 for end-point 2 or if end-point 1 nonsignificant followed by P ≤ 0.025 for end-point 2. End-point 1 can be examined in Step 3 if deemed nonsignificant in Step 1. The P value should be ≤0.05 preceded by a significant treatment effect over end-point 2 in Step 2 |
Sometimes advanced gate keeping tests like Hommels' test are necessary in trials having two or three sources of multiplicity, as in the case of lurasidoneprogramme in schizophrenia[7],[10] [Figure - 3]. In this study with several primary or secondary objectives, multiple dose-placebo comparisons along with other factors had prompted complex multiplicity issues. Such strategies are being increasingly used in complex phase III trials with several sources of multiplicity.[13]
 |
| Figure 3: Predicted testing pathways in a Phase III trial of lurasidone versus placebo comparing two doses in three outcome parameters |
Multiplicity Adjustment in Subpopulation Analysis: Example of Apex Study
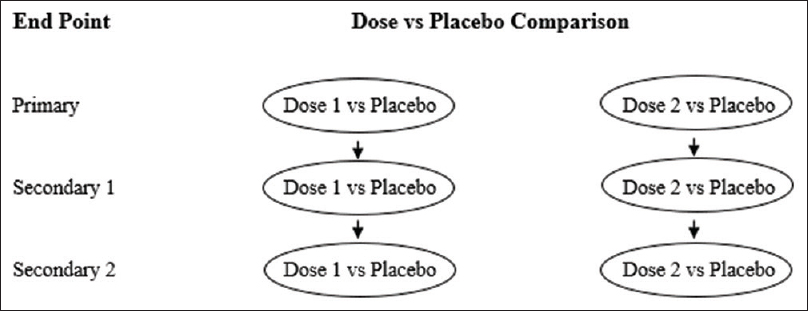
In recent years, the trend of “targeted” therapies has led to complex phase III trials based upon multiple-population analysis. Increasingly, the efficacy of new treatment is being analyzed in different subgroups, including the intention-to-treat population or all-comers population.[14] For instance, in the APEX trial, which examined the advantages of betrixaban over enoxaparin in patients at risk of venous thrombosis, the primary analysis was done in AP and two different subpopulations (S1 and S2) [Figure - 4] and [Figure - 5].[5] The two-sided P value for between-group difference in study population is depicted in [Table - 1]. The flexible and adaptable decision path in Hochberg's test [as depicted in [Figure - 5] would assist the trial sponsor to enable efficacy claim over one patient population even if the treatment effect is nonsignificant over another subpopulation.[10]
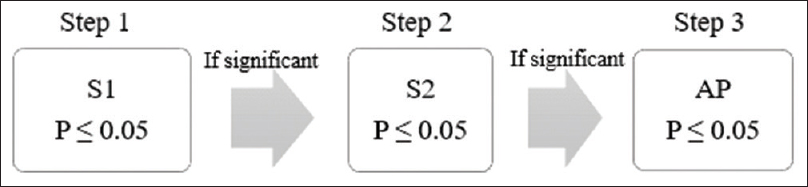 |
| Figure 4: Simulation of fixed sequence testing strategy in APEX trial: Significance level should be tested at S1 followed by S2 and lastly at AP. Treatment effect over S1 is deemed significant at P ≤ 0.05. In Step 2, treatment effect over S2 is deemed significant if Step 1 is significant followed by P ≤ 0.05 in Step 2. Subsequently, treatment effect over AP in Step 3 is conferred significant if Step 2 is significant followed by P ≤ 0.05 in Step 3 |
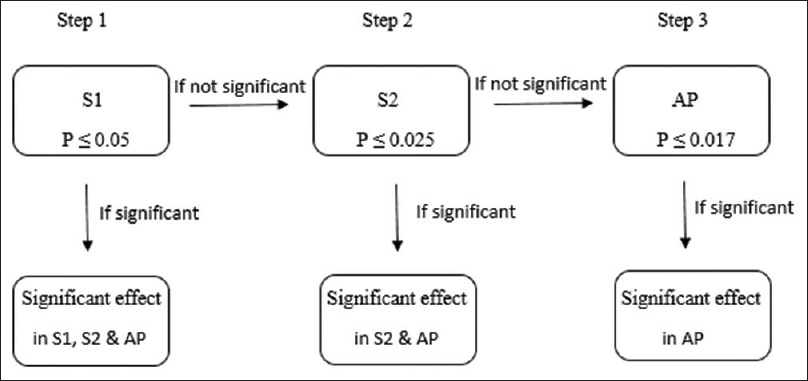 |
| Figure 5: Simulation of Hochberg's testing strategy in APEX trial: S1 has to be tested first as it corresponds to the largest P value followed by S2 and AP. In Step 1, treatment effect over S1, S2, and AP is deemed significant at P ≤ 0.05. In Step 2, treatment effect over S2 and AP is deemed significant if treatment effect was nonsignificant in Step 1 but P ≤ 0.025 in Step 2. Treatment effect is significant in AP if treatment effect was deemed nonsignificant in Step 2 but P ≤ 0.017 in Step 3 |

Multiplicity Adjustment in Dermatological Trials
The CIMPASI-1 and CIMPASI-2 studies were designed to assess the efficacy and safety of Certolizumab Pegol (an Fc-free, PEGylated antitumor necrosis factor biologic) in moderate-to-severe chronic plaque psoriasis. Subjects were randomized 2:2:1 to certolizumab 400 mg, certolizumab 200 mg, or placebo every 2 weeks.[15],[16] The coprimary end-points were week 16 responder rates, defined as a 75% reduction in Psoriasis Area and Severity Index and Physician's Global Assessment (PGA) 0/1 (clear/almost clear) and ≥2-point improvement. Safety was assessed by treatment-emergent adverse events. In the pooled analysis of ongoing CIMPASI studies, multiplicity was controlled by fixed sequence testing procedure.[15]Bonferroni method was applied to the overall significance level of 0.05 to divide it by 2, that is, 0.025 to each dose (Certolizumab Pegol 400 and 200 mg). The hypothesis was arranged in two sets [H1, H3, H5, H7; H2,H4, H6, H8] as each hypothesis within a set represented the same dose for a specified outcome, while the alpha was split equally [Figure - 6]. Interestingly, Hommel's test could have been used (though not clarified in the trial), to check for multiplicity attributed to multiple dose–outcome relationship in comparison to the placebo.
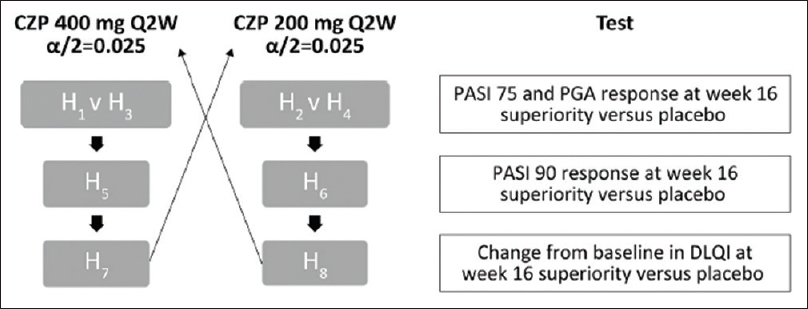 |
| Figure 6: Fixed-sequence testing procedure, PASI 75, ≥75% reduction in PASI from baseline PASI; PASI 90, ≥90% reduction in PASI from baseline PASI; PGA Q2W, every 2 weeks. Reproduced from Gottlieb et al. CZP: Certolizumab Pegol, DLQI: Dermatology Life Quality Index, PASI: Psoriasis Area and Severity Index, PGA: Physician's Global Assessment |
There are other studies depicting adjustments for multiplicity such as RESTORE1 trial, in which efficacy and safety of infliximab versus methotrexate was assessed in patients with moderate-to-severe plaque psoriasis.[17] The major secondary efficacy end-points were PASI 75 response at week 26 and the proportion of patients achieving a PGA score of 0 (cleared) or 1 (minimal) at weeks 16 and 26. All the secondary end points were adjusted using the Hochberg test. As previously mentioned, it allows more flexible way of testing than the fixed-sequencetesting procedures in the subpopulation analysis. This test can also be used for multiplicity adjustments for secondary end-points as specified in RESTORE1 trial. All the major secondary end-points were deemed to be significantly improved in the infliximab group.
The flexible decision path adopted by Hochberg test was also used in a trial of a 12-week course of efalizumab 1 mg/kg subcutaneous compared with placebo. There was a significant improvement in the efficacy parameters such as the proportion of patients achieving PASI-75 and PASI-50, sPGA rating of minimal or clear, and the change in patients' Psoriatic Symptom Assessment (PSA) from baseline after multiplicity adjustment.[18]
On the contrary, in ESTEEM-2 trial the efficacy and safety of apremilast, an oral PDE-4 inhibitor, was assessed using a hierarchical or fixed sequence testing for multiplicity adjustments for secondary end-points.[19] The major secondary end-point of the proportion of patients achieving sPGA response at week 16 was significantly improved compared with placebo.
Thus, the examples show that in principle, trials in dermatology are no different from the trials done in other specialities. In trials where multiple doses of a testing agent are looked for primary and secondary outcomes, the issue of multiplicity may arise if not properly adjusted. This might become more complex if tested in different subpopulations. However, a recent systematic review of randomized controlled trials in different dermatology journals has raised a definite apprehension regarding the low reporting of statistical variables in different studies, including paucity in multiplicity adjustment.[20] There were very few studies using methods such as Bonferroni's, Holm's, and Dunn's with a higher tendency of wrong rejection of the null hypothesis. It necessitates intervention to increase the level of statistical reporting in dermatological studies to increase the validity and reliability.
Multiplicity Adjustment in Interim Analysis
Interim analysis is the key to the monitoring of a lengthy complex trial in prespecified time point attributed to the termination of a trial in the verge of confirmed benefit or unexpected harm. There are diverse prospective statistical strategies for stopping a clinical trial early. Overall, the stopping rule for interim analysis needs to be conservative with respect to using more stringent P values to achieve significance level close to 0.05 in the final analysis.[1] For instance, using of P value ≤0.00003 during the first half and P value ≤0.002 in the second half of the HOPE (Heart Outcomes Prevention Evaluation) trial, retained the P value close to 0.05 for final analysis.[21] Alternatively, the stopping rule can be based on the estimate of treatment effect (O'Brien and Fleming's method, Kittleson and Emerson's method), the normalised Z-statistics, the fixed sample P value (Pocock's method), and error spending function (Lan and Demet's approach and Kim and DeMet's method).[22]
Comments
The US FDA and European Medicines Agency (EMA) have recently published a draft document on multiplicity in a clinical trial.[2],[3] The document highlighted the principle and basis of multiple comparisons arising in a confirmatory clinical trial with multiple objectives. However, there has been a debate over addressing multiplicity issues in exploratory or early phase trials, an issue that has not been satisfactorily explained in the draft guidelines. As of now, the trial sponsor needs to avert incorrect statistical interpretation in the initial trial phases, using robust statistical techniques. Examples include the MCP-Mod algorithm, a dose-finding strategy in phase II taking multiplicity adjustment into account.[23]
The analytical strategy of multiplicity adjustments has been promulgated as the trial complexity has attained a high over the past few years. The basic tenets are to address different issues of multiplicity avoiding spurious finding in trial results. Future confirmatory studies are likely to be structured on several end points to attain multiple clinical objectives. Therefore, it is essential to consider all relevant statistical and clinical information to make a comprehended strategy in accordance with the trial objectives. It must include information about end-points of interest, different subpopulation, and other key statistical features.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Li G, Taljaard M, Van den Heuvel ER, Levine MA, Cook DJ, Wells GA, et al. An introduction to multiplicity issues in clinical trials: The what, why, when and how. Int J Epidemiol 2017;46:746-55.
[Google Scholar]
|
| 2. |
Center for Drug Evaluation and Research. Multiple Endpoints in Clinical Trials Guidance for Industry. Center for Drug Evaluation and Research; 2017. Available from: http://www.fda.gov/downloads/ drugs/guidancecomplianceregulatoryinformation/guidances/ ucm536750.pdf. [Last retrieved on 2018 Dec 12].
[Google Scholar]
|
| 3. |
Draft Guideline on Multiplicity Issues in Clinical Trials (EMA/ CHMP/44762/2017). Committee for Human Medicinal Products; 2016. Available from: http://www.ema.europa.eu/documents/scientific-guideline/draft-guideline-multiplicity-issues-clinical-trials_en.pdf. [Last retrieved on 2018 Dec 12].
[Google Scholar]
|
| 4. |
Dmitrienko A, D'Agostino RB Sr. Editorial: Multiplicity issues in clinical trials. Stat Med 2017; 36:4423-6.
[Google Scholar]
|
| 5. |
Cohen AT, Harrington RA, Goldhaber SZ, Hull RD, Wiens BL, Gold A, et al. Extended thromboprophylaxis with betrixaban in acutely ill medical patients. N Engl J Med 2016;375:534-44.
[Google Scholar]
|
| 6. |
Rosenstock J, Aguilar-Salinas C, Klein E, Nepal S, List J, Chen R, et al. Effect of saxagliptin monotherapy in treatment-naïve patients with type 2 diabetes. Curr Med Res Opin 2009;25:2401-11.
[Google Scholar]
|
| 7. |
Nasrallah HA, Silva R, Phillips D, Cucchiaro J, Hsu J, Xu J, et al. Lurasidone for the treatment of acutely psychotic patients with schizophrenia: A 6-week, randomized, placebo-controlled study. J Psychiatr Res 2013;47:670-7.
[Google Scholar]
|
| 8. |
PI3K inhibitor improves PFS in BELLE-2 trial. Cancer Discov 2016;6:115-6.
[Google Scholar]
|
| 9. |
Dmitrienko A, D'Agostino RB Sr. Multiplicity considerations in clinical trials. N Engl J Med 2018; 378:2115-22.
[Google Scholar]
|
| 10. |
Dmitrienko A, D'Agostino RB Sr., Huque MF. Key multiplicity issues in clinical drug development. Stat Med 2013; 32:1079-111.
[Google Scholar]
|
| 11. |
Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368:138-48.
[Google Scholar]
|
| 12. |
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371:424-33.
[Google Scholar]
|
| 13. |
Brechenmacher T, Xu J, Dmitrienko A, Tamhane AC. A mixture gatekeeping procedure based on the Hommel test for clinical trial applications. J Biopharm Stat 2011;21:748-67.
[Google Scholar]
|
| 14. |
Millen BA, Dmitrienko A, Ruberg S, Shen Ll. A statistical framework for decision making in confirmatory multipopulation tailoring clinical trials. Drug Inform J 2012;46:647-56.
[Google Scholar]
|
| 15. |
Gottlieb AB, Blauvelt A, Thaçi D, Leonardi CL, Poulin Y, Drew J, et al. Certolizumab pegol for the treatment of chronic plaque psoriasis: Results through 48 weeks from 2 phase 3, multicenter, randomized, double-blinded, placebo-controlled studies (CIMPASI-1 and CIMPASI-2). J Am Acad Dermatol 2018;79:302-314.e6.
[Google Scholar]
|
| 16. |
Blauvelt A, Reich K, Lebwohl M, Burge D, Arendt C, Peterson L, et al. Certolizumab pegol for the treatment of patients with moderate-to-severe chronic plaque psoriasis: Pooled analysis of week 16 data from three randomized controlled trials. J Eur Acad Dermatol Venereol 2019;33:546-52.
[Google Scholar]
|
| 17. |
Barker J, Hoffmann M, Wozel G, Ortonne JP, Zheng H, van Hoogstraten H, et al. Efficacy and safety of infliximab vs. methotrexate in patients with moderate-to-severe plaque psoriasis: Results of an open-label, active-controlled, randomized trial (RESTORE1). Br J Dermatol 2011; 165:1109-17.
[Google Scholar]
|
| 18. |
Papp KA, Bressinck R, Fretzin S, Goffe B, Kempers S, Gordon KB, et al. Safety of efalizumab in adults with chronic moderate to severe plaque psoriasis: A phase IIIb, randomized, controlled trial. Int J Dermatol 2006; 45:605-14.
[Google Scholar]
|
| 19. |
Paul C, Cather J, Gooderham M, Poulin Y, Mrowietz U, Ferrandiz C, et al. Efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with moderate-to-severe plaque psoriasis over 52 weeks: A phase III, randomized controlled trial (ESTEEM 2). Br J Dermatol 2015; 173:1387-99.
[Google Scholar]
|
| 20. |
McClean M, Silverberg JI. Statistical reporting in randomized controlled trials from the dermatology literature: A review of 44 dermatology journals. Br J Dermatol 2015; 173:172-83.
[Google Scholar]
|
| 21. |
Heart Outcomes Prevention Evaluation Study Investigators, Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med 2000; 342:145-53.
[Google Scholar]
|
| 22. |
Emerson SS, Kittelson JM, Gillen DL. Frequentist evaluation of group sequential clinical trial designs. Stat Med 2007; 26:5047-80.
[Google Scholar]
|
| 23. |
Bretz F, Pinheiro JC, Branson M. Combining multiple comparisons and modeling techniques in dose-response studies. Biometrics 2005; 61:738-48.
[Google Scholar]
|
Fulltext Views
4,569
PDF downloads
4,239





![[Figure - 1]](#fig_ijdvl_2020_86_2_222_277254_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_2_222_277254_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2020_86_2_222_277254_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2020_86_2_222_277254_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2020_86_2_222_277254_f5.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2020_86_2_222_277254_t7.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2020_86_2_222_277254_f6.jpg){kind=link}