Translate this page into:
Ectodermal dysplasia-skin fragility syndrome with a new mutation
2 Department of Dermatology, Mugla Sitki Kocman University Training and Research Hospital, Mugla, Turkey
3 Department of Medical Genetics, Faculty of Medicine, Mugla Sitki Kocman University, Mugla, Turkey
4 Department of Molecular Biology and Genetic, Faculty of Life Sciences, Mugla Sitki Kocman University, Mugla, Turkey
Correspondence Address:
Emine Tugba Alatas
Department of Dermatology, Mugla Sitki Kocman University, Faculty of Medicine, 48000 Mugla
Turkey
| How to cite this article: Alatas ET, Kara A, Kara M, Dogan G, Baysal O. Ectodermal dysplasia-skin fragility syndrome with a new mutation. Indian J Dermatol Venereol Leprol 2017;83:476-479 |
Sir,
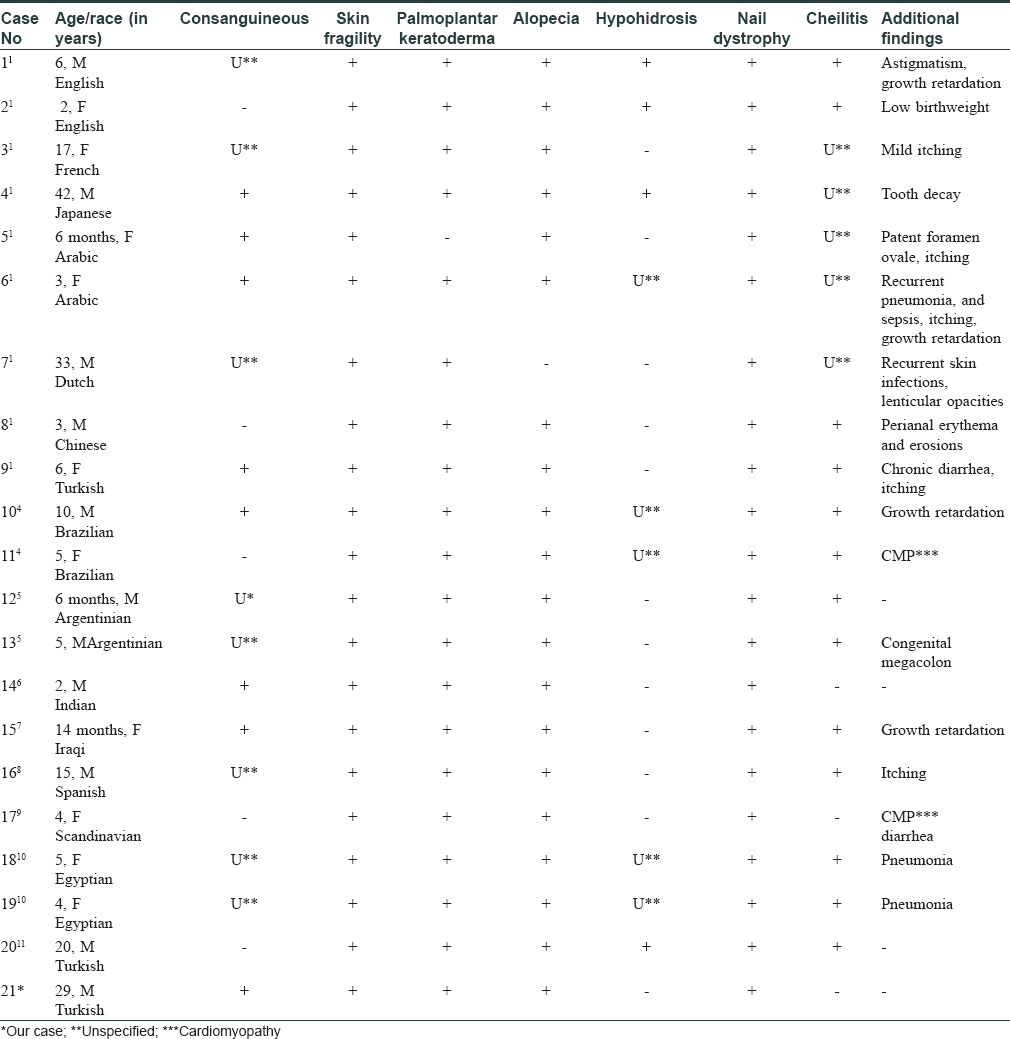
Ectodermal dysplasia-skin fragility syndrome (EDDFS) was first described in 1997 by Mcgrath et al., in a 6-year-old British boy with skin fragility, short sparse hair, erosions and blistering.[1] EDDFS occurs as a result of mutation in the PKP 1 gene, and is characterized by widespread skin fragility, alopecia, nail dystrophy, palmoplantar keratoderma and painful fissures.[1],[2],[3],[4] In literature, we could find about twenty cases [Table - 1] describing this entity [2],[5],[6],[7],[8],[9],[10],[11],[12] and two of them are from Turkey.
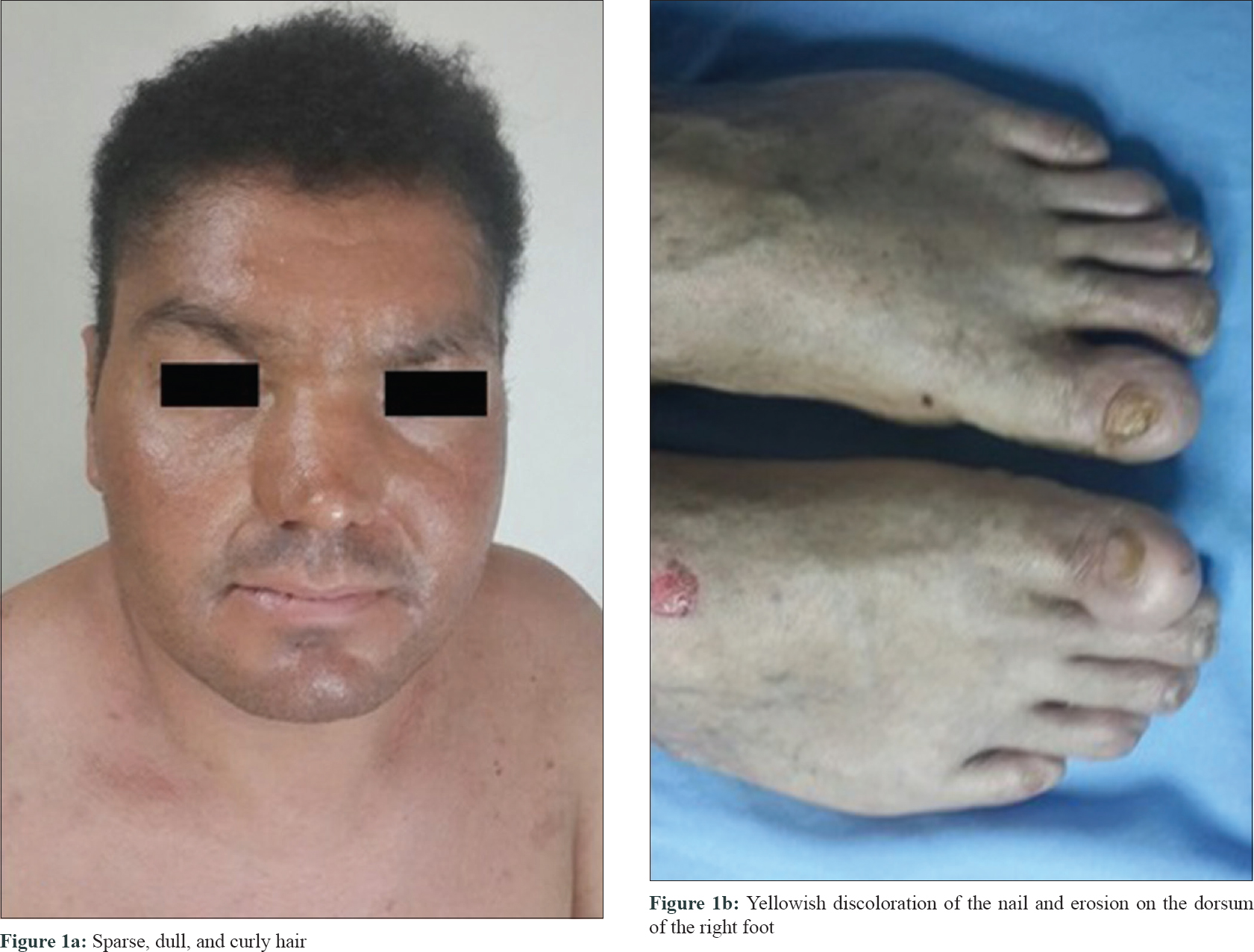
A 29-year-old male patient was admitted with complaints of thickening and fissuring of the skin of the soles. His complaints were present since birth and he also gave a history of reduced sweating. His parents were second-degree relatives and his sister reportedly died at the age of 5 years with similar complaints. His parents and three brothers were asymptomatic.
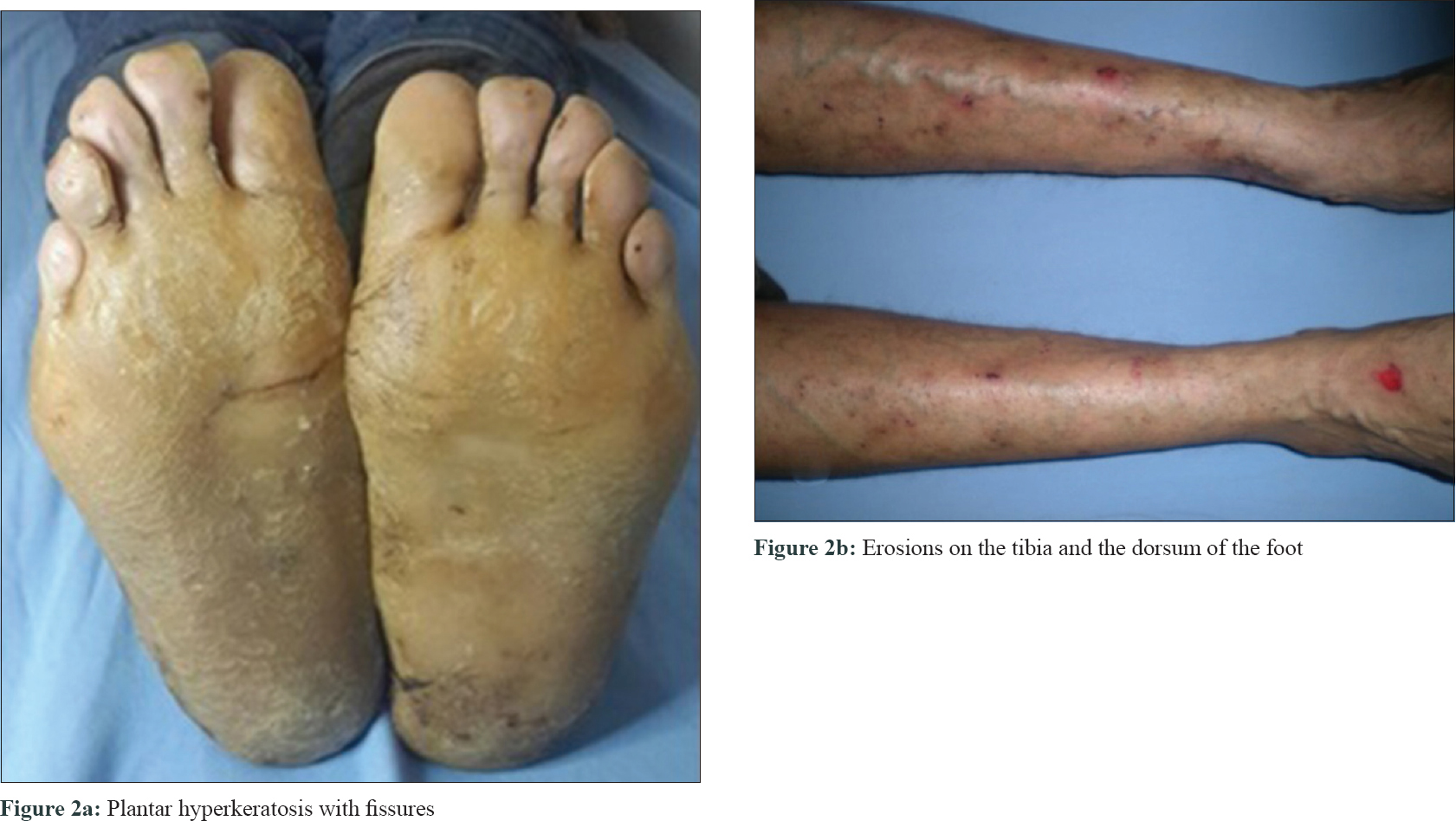
Dermatological examination revealed diffuse keratoses and deep fissures in the palmoplantar regions. Erosions were seen on dorsal surface of the hands. The patient had a flat nasal bridge. His scalp was normal, but his hair appeared short, curly, thin and brittle. The hair pull test revealed easily broken hairs [Figure - 1] and [Figure - 2]. Dystrophic changes were seen in the nails as well. Histopathological examination revealed diffuse parakeratosis, prominent granular layer, submucosal cleavage in the focal area and increased intercellular edema between the keratinocytes.
 |
| Figure 1 |
 |
| Figure 2 |
DNA sequencing analysis was done which detected a novel heterozygous p. Val472Glyfs*28 (c.1414_1415delTG) mutation in our index case, and in the father and mother of the case. This mutation, which has not been previously reported, leads to premature protein termination. Two bioinformatics tools - PolyPhen-2 and Mutation Taster, predict the p. Val472Glyfs*28 (c.1414_1415delTG) change to be, “probably damaging”, and “disease causing”, respectively [Figure - 3]. Ectodermal dysplasia-skin fragility syndrome was diagnosed with these findings.
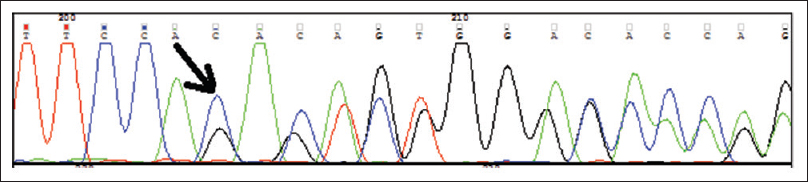 |
| Figure 3: DNA sequencing analysis |
Ectodermal dysplasia-skin fragility syndrome is a rare inherited autosomal recessive genodermatosis that is recently defined. EDDFS is characterized by PKP 1 mutation, which results in the characteristic clinical findings in the skin, hair and nails.[1] It was learned that the patient's sister died with similar symptoms when she was five years old. His mother, father and brothers had no related complaints.
PKP1 is a component of the desmosomal plaque. This protein contributes to the mechanical integrity of the desmosomes by binding desmosomal cadherins to keratin.[3] PKP 1 protein is encoded by the PKP 1 gene. This gene is located, especially in the supra-basal cells and the outer layers of the hair follicles in the epidermis. EDDFS occurs as a result of loss of function mutations of this gene which result in changes in PKP 1 protein levels.[2] Blisters caused by trauma are specific findings of epidermolysis bullosa (EB) disease. Therefore, this syndrome is classified as a specific suprabasal form of EB simplex at the IIIrd International Consensus Meeting for the diagnosis and classification of EB.[4]
EDDFS was first described by McGrath et al. in 1997.[1] Some specific signs occur in the skin, hair and nails. Fragility and bullous lesions secondary to trauma can be commonly seen in EDDFS. Also, erosions, palmoplantar keratoderma and fissures secondary to keratoses are common. Alopecia or thinning hair which is dry, short and curly is the typical appearence. Dystrophic changes in the nail are also frequent findings.[1] Hypohidrosis, pruritus, perioral fissures, cheilits, growth retardation, and other aberrations are rare clinical findings [Table - 1].[2],[5],[6],[7],[8],[9],[10],[11],[12]
Hypohidrosis has been reported in a 40-year-old Japanese patient and a 20-year-old male Turkish patient.[2],[12] Cheilitis has been observed in a 2-year-old Indian male patient, and 4-year-old Scandinavian woman.[7],[10] In our case, there were palmoplantar keratoderma with erosions of the skin, brittle hair and nail dystrophy. EDDFS may also be associated with cardiomyopathy, patent foramen ovale, chronic diarrhea and congenital megacolon. We did not observe any pathological findings in electrocardiogram, chest X-ray, pulmonary function tests and echocardiogram examinations. We also did not detect any other co-morbidity.
Ersoy et al. has reported EDDFS resulting from the homozygous mutation, 888delC, in the desmosomal protein plakophilin 1. Whittock et al. found a homozygous splice site mutation (1233–2 A→T; GenBank Z73678) in a 17 year-old male with EDDFS.[2] Sequencing of the genomic DNA revealed a homozygous 5 base pair deletion in exon 5 of the PKP1 gene, designated as c.897del5 (CAACC).[8] We found p. Val472Glyfs*28 (c.1414_1415delTG) mutation in our case, our case's father and the mother, unlike in other reports.
Treatment of EEDFS is generally symptomatic. Wound care and the diagnosis and treatment of secondary bacterial infections are important to reduce morbidity.[2]
We are reporting this case due to the rarity in the literature. The detection of a new mutation for this disease makes it more interesting and adds to the present knowledge and understanding about the disease.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient has given his consent for his images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
McGrath JA, McMillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, et al. Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet 1997;17:240-4.
[Google Scholar]
|
| 2. |
Ersoy-Evans S, Erkin G, Fassihi H, Chan I, Paller AS, Surucu S,et al. Ectodermal dysplasia-skin fragility syndrome resulting from a new homozygous mutation, 888delC, in the desmosomal protein plakophilin1. J Am Acad Dermatol 2006;55:157-61.
[Google Scholar]
|
| 3. |
Schmidt A, Jager S. Plakophilins - hard work in the desmosome, recreation in the nucleus?. European Journal of Cell Biology 2005;84:189-204.
[Google Scholar]
|
| 4. |
Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the third International Consensus Meeting on diagnosis and classification of EB. J Am Acad Dermatol 2008;58:931-50.
[Google Scholar]
|
| 5. |
Tanaka A, Lai-Cheong JE, Café ME, Gontijo B, Salomão PR, Pereira L, et al. Novel truncating mutations in PKP1 and DSP cause similar skin phenotypes in two Brazilian families. Br J Dermatol 2009;160:692-7.
[Google Scholar]
|
| 6. |
Winik BC, Asial RA, McGrath JA, South AP, Boente MC. Acantholytic ectodermal dysplasia: Clinicopathological study of a new desmosomal disorder. Br J Dermatol 2009;160:868-74.
[Google Scholar]
|
| 7. |
Adhe VS, Dongre AM, Khopkar US. Ectodermal dysplasia-skin fragility syndrome. Indian J Dermatol Venereol Leprol 2011;77:503-6.
[Google Scholar]
|
| 8. |
Boyce AE, McGrath JA, Techanukul T, Murrell DF, Chow CW, McGregor L, et al. Ectodermal dysplasia-skin fragility syndrome due to a new homozygous internal deletion mutation in the PKP1 gene. Australas J Dermatol 2012;53:61-5.
[Google Scholar]
|
| 9. |
Hernández-Martín A, Torrelo A, Ciria S, Colmenero I, Aguilar A, Grimalt R, et al. Ectodermal dysplasia-skin fragility syndrome: A novel mutation in the PKP1 gene. Clin Exp Dermatol 2013;38:787-90.
[Google Scholar]
|
| 10. |
Vahlquist A, Virtanen M, Hellström-Pigg M, Dragomir A, Ryberg K, Wilson NJ, et al. A Scandinavian case of skin fragility, alopecia and cardiomyopathy caused by DSP mutations. Clin Exp Dermatol 2014;39:30-4.
[Google Scholar]
|
| 11. |
Abdalla EM, Has C.A plakophilin-1 gene mutation in an egyptian family with ectodermal dysplasia-skin fragility syndrome. Mol Syndromol 2014;5:304-6.
[Google Scholar]
|
| 12. |
Atalay AA, Fatos O, Abuaf K. Ektodermal Displazi Deri Frajilite Sendromu. Turk J Dermatol 2014; 2:114-7.
[Google Scholar]
|
Fulltext Views
4,155
PDF downloads
2,267





![[Table - 1]](#tbl_ijdvl_2017_83_4_476_206381_t2.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2017_83_4_476_206381_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2017_83_4_476_206381_f3.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2017_83_4_476_206381_f4.jpg){kind=link}