Translate this page into:
Epidermolysis bullosa pruriginosa: A case with a novel mutation and co-existent lichen amyloidosus
Correspondence Address:
Qiping Chen
National Skin Centre, 1 Mandalay Road Singapore - 308205
Singapore
| How to cite this article: Chen Q, Lee JS, Tey HL. Epidermolysis bullosa pruriginosa: A case with a novel mutation and co-existent lichen amyloidosus. Indian J Dermatol Venereol Leprol 2015;81:40-42 |
Abstract
Epidermolysis bullosa pruriginosa is a rare variant of dystrophic epidermolysis bullosa characterized by severely pruritic and cicatricial lesions localized to the extensor extremities. We report a Singaporean Chinese male with epidermolysis bullosa pruriginosa with an underlying novel mutation in the COL7A1 gene. A heterozygous acceptor splice site mutation IVS67-1G>T probably led to in-frame skipping of exon 68 (36-basepairs), resulting in a loss of 12 amino acids. Among his three children, only the youngest son, who had bilateral big toenail thickening, possessed the same mutation. His skin biopsy one decade ago revealed association of focal amyloidosis; a recent skin biopsy showed more established features of lichen amyloidosis. It is debatable whether the cutaneous amyloidosis was a secondary or primary phenomenon. Our report highlights that the diagnosis of epidermolysis bullosa pruriginosa may be obscured when cutaneous amyloidosis is coexistent.INTRODUCTION
Epidermolysis bullosa pruriginosa is a rare variant of autosomal dominant (and occasionally autosomal recessive) dystrophic epidermolysis bullosa. [1] Mutations in the COL7A1 gene, which encode type VII collagen, especially in substitution of glycine, have been shown to be the cause. It results in anchoring fibril dysfunction at the dermoepidermal junction and blistering beneath the lamina densa of the epidermal basement membrane. [2] In addition to the features of dystrophic epidermolysis bullosa which include trauma-induced blisters, milia, scars, and nail dystrophy, epidermolysis bullosa pruriginosa is characterized by a delayed age of onset, severe localized or generalized pruritus, scratching-induced secondary skin lesions and pronounced scarring localized predominantly to the extensor surface of the extremities. We report a patient with epidermolysis bullosa pruriginosa in association with lichen amyloidosis.
CASE REPORT
A 53-year-old Singaporean Chinese first presented to the National Skin Centre, Singapore, 14 years ago with severely pruritic rashes on the lower limbs. The symptom started since he was in his late 30s and led him to scratch frequently, resulting in blisters and erosions. On examination, there were multiple keratotic and dermal papules on the lower legs which were associated with erosions and milia [[Figure - 1]a and b]. He also had nail dystrophy involving multiple finger and toe nails. A skin biopsy revealed the presence of epidermal acanthosis, subepidermal clefting, milia formation, and dermal scarring with regenerating blood vessels [[Figure - 2]a], consistent with features of epidermolysis bullosa pruriginosa. In addition, there was focal lichen amyloidosis [[Figure - 2]b], consisting of Congo red-positive deposits of amyloid within the upper dermis. Over the years, the papules gradually became more keratotic and firm. The clinical picture suggested lichen amyloidosis and a recent repeat skin biopsy when he was 68 years old revealed a more florid picture of lichen amyloidosus.
 |
| Figure 1: (a) Multiple keratotic and dermal papules on the lower legs. (b) Close-up view revealed the presence of milia (straight arrows) and excoriations/erosions (curved arrows) |
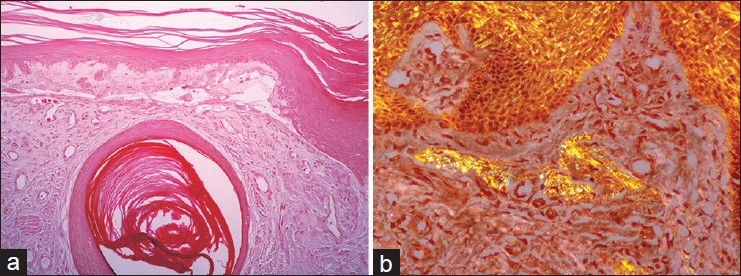 |
| Figure 2: (a) There was a subepidermal blister with a milium and increased small dermal blood vessels within a fibrotic stroma. The epidermis showed mild acanthosis (Hematoxylin and Eosin, ×100). (b) There were aggregates of amyloid within the papillary dermis which stain positively for Congo Red, and showed apple green birefringence on polarized microscopy (Congo red stain on polarized microscopy, ×200) |
Gene sequencing for mutation in the COL7A gene was performed and a novel heterozygous mutation IVS67-1G>T was found. This is an acceptor splice site mutation occurring in the area of the gene encoding the triple helix of type VII collagen. There was no family history of similar involvement except that his youngest son had bilateral big toe nail dystrophy. Gene sequencing at the site of the patient′s mutation in the COL7A1 gene was performed for all his three children. Among them, only the youngest son possessed the same heterozygous mutation, reflecting dominant inheritance of the condition.
Blood investigations including full blood count, renal function, liver function and human immunodeficiency virus screening for the patient did not reveal any abnormalities. The patient was treated with potent topical steroids, 0.1% tacrolimus ointment and soak PUVA; however, these were not efficacious in reducing his itch or skin lesions.
DISCUSSION
Epidermolysis bullosa pruriginosa is a severely pruritic inherited disease which can be easily misdiagnosed due to its rarity, late age of onset and secondary cutaneous changes. The typical skin lesions, consisting of keratotic plaques associated with deep excoriations, points to severe scratching, and this frequently leads to misdiagnosis of acquired disorders such as dermatitis artefacta, nodular prurigo, lichen simplex chronicus, pemphigoid nodularis, and hypertrophic lichen planus. In our patient, the genetic mutation in the COL7A1 gene, together with supportive histological findings, clinches the diagnosis of epidermolysis bullosa pruriginosa.
In the literature, more than 40 different mutations in the COL7A1 gene have been reported in epidermolysis bullosa pruriginosa so far, including missense, splice-site mutations and small nucleotide deletions. [3],[4],[5],[6],[7],[8],[9] Comparing dystrophic epidermolysis bullosa and epidermolysis bullosa pruriginosa, there is generally no clear genotype-phenotype correlation although skipping of exon 87 in the COL7A1 gene can be associated with the dominantly-inherited form of epidermolysis bullosa pruriginosa. [7],[8],[9] The novel dominant-negative heterozygous acceptor splice site mutation in the COL7A1 gene (IVS67-1G>T) was found in both our patient and his youngest son, who was 34 years old at the time of testing. The latter can reasonably be assumed to be at risk of developing epidermolysis bullosa pruriginosa in the future and should receive treatment as soon as symptoms arise. Genetic counseling had also been performed. This particular mutation has not been reported in epidermolysis bullosa pruriginosa before. It represents a splice site mutation, a number of which have been previously shown to be pathogenic in both dominant and recessive forms of dystrophic epidermolysis bullosa. This particular mutation probably led to in-frame skipping of exon 68 (36-bp), resulting in a loss of 12 amino acids. This could then lead to dominant-negative interference between wild-type and truncated type VII collagen protein, i.e., a dominant form of dystrophic epidermolysis bullosa. This molecular finding is consistent with the clinical phenotype of epidermolysis bullosa pruriginosa.
We were unable to find any previous reports of the association of epidermolysis bullosa pruriginosa with localized cutaneous amyloidosis. Although there are a few reports of the association of epidermolysis bullosa, especially recessive epidermolysis bullosa, with systemic amyloidosis, only one case of pretibial dystrophic epidermolysis bullosa with localized cutaneous amyloidosis has been reported so far. [10] The cutaneous amyloidosis could be a primary unrelated event, or secondary to chronic scratching from pruritus associated with epidermolysis bullosa pruriginosa. In view of the clinical course, we favor the lichen amyloidosis occurring as a secondary event to chronic scratching. However, primary lichen amyloidosis is not uncommon in older individuals of Southern Chinese heritage and both epidermolysis bullosa pruriginosa and lichen amyloidosis typically occur on the shins; therefore the co-existence of both conditions could be unrelated. As both epidermolysis bullosa pruriginosa and lichen amyloidosis are associated with a high degree of itch, the presence of both conditions is likely to intensify the degree of pruritus and worsen the itch-scratch cycle.
In summary, we report a case of epidermolysis bullosa pruriginosa with a novel splice-site mutation (IVS67-1G>T) in the COL7A1 gene and co-existent lichen amyloidosis. The diagnosis of epidermolysis bullosa pruriginosa is challenging, particularly with the presence of lichen amyloidosis, as in this case.
| 1. |
McGrath JA, Schofield OM, Eady RA. Epidermolysis bullosa pruriginosa: Dystrophic epidermolysis bullosa with distinctive clinicopathological features. Br J Dermatol 1994;130:617-25.
[Google Scholar]
|
| 2. |
Lee JY, Pulkkinen L, Liu HS, Chen YF, Uitto J. A glycine-to-arginine substitution in the triple-helical domain of type VII collagen in a family with dominant dystrophic epidermolysis bullosa pruriginosa. J Invest Dermatol 1997;108:947-9.
[Google Scholar]
|
| 3. |
Mellerio JE, Ashton GH, Mohammedi R, Lyon CC, Kirby B, Harman KE, et al. Allelic heterogeneity of dominant and recessive COL7A1 mutations underlying epidermolysis bullosa pruriginosa. J Invest Dermatol 1999;112:984-7.
[Google Scholar]
|
| 4. |
Ee HL, Liu L, Goh CL, McGrath JA. Clinical and molecular dilemmas in the diagnosis of familial epidermolysis bullosa pruriginosa. J Am Acad Dermatol 2007;56:S77-81.
[Google Scholar]
|
| 5. |
Tey HL, Lee AD, Almaani N, McGrath JA, Mills KC, Yosipovitch G. Epidermolysis bullosa pruriginosa masquerading as psychogenic pruritus. Arch Dermatol 2011;147:956-60.
[Google Scholar]
|
| 6. |
Saito M, Masunaga T, Ishiko A. A novel de novo splice-site mutation in the COL7A1 gene in dominant dystrophic epidermolysis bullosa (DDEB): Specific exon skipping could be a prognostic factor for DDEB pruriginosa. Clin Exp Dermatol 2009;34:e934-6.
[Google Scholar]
|
| 7. |
Jiang W, Sun TT, Lei PC, Zhu XJ. Genotype-phenotype correlation in Chinese patients with dystrophic epidermolysis bullosa pruriginosa. Acta Derm Venereol 2012;92:50-3.
[Google Scholar]
|
| 8. |
Fortuna G, Di Lorenzo M, Cepeda-Valdes R, Garcia-Garcia C, Salas-Alanis JC. The largest family of the Americas with dominant dystrophic epidermolysis bullosa pruriginosa: A 18-year longitudinal genotype-phenotype study. J Dermatol Sci 2013;71:217-21.
[Google Scholar]
|
| 9. |
Tang ZL, Lin ZM, Wang HJ, Chen Q, Xu XM, Ge HF, et al. Four novel and two recurrent glycine substitution mutations in the COL7A1 gene in Chinese patients with epidermolysis bullosa pruriginosa. Clin Exp Dermatol 2013;38:197-9.
[Google Scholar]
|
| 10. |
Aoki M, Niimi Y, Ishiko A, Kawana S. Pretibial dystrophic epidermolysis bullosa with localized cutaneous amyloidosis: Coincidental or secondary amyloidosis? J Dermatol 2010;37:259-63.
[Google Scholar]
|
Fulltext Views
2,828
PDF downloads
1,668





![[Figure - 1]](#fig_ijdvl_2015_81_1_40_148565_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_1_40_148565_f2.jpg){kind=link}