Translate this page into:
Cutaneous T-cell non Hodgkin lymphoma in a patient with idiopathic CD4+ lymphocytopenia
Correspondence Address:
Sarita Sasidharanpillai
'Rohini', Girish Nagar, Nallalom PO, Kozhikode - 673 027, Kerala
India
| How to cite this article: Riyaz N, Sasidharanpillai S, Khader A, Puravoor J. Cutaneous T-cell non Hodgkin lymphoma in a patient with idiopathic CD4+ lymphocytopenia. Indian J Dermatol Venereol Leprol 2013;79:831-833 |
Sir,
Idiopathic CD4+ lymphocytopenia (ICL) is a rare heterogenous condition, often detected in middle age, following the occurrence of an opportunistic infection in a person without known immunodeficiency or immunosuppression. [1] Here we report, a patient with ICL who presented with recurrent furuncles and who subsequently developed cutaneous T-cell non-Hodgkin lymphoma (T-cell NHL).
A 33-year-old man attended the out-patient-department of our hospital with an 8 month history of recurrent furuncles distributed over scalp, lower limbs, and back of trunk. His past medical history was insignificant and he was not on any drugs. Swab culture from furuncles confirmed staphylococcal infection.
A detailed evaluation for any underlying immunosuppression including a complete hemogram, liver, and renal function tests, random blood sugar, serum immunoglobulins, chest X-ray, and peripheral smear were within normal limits. Serology for p24 antigen, HIV 1 and 2 and human T cell leukemia virus (HTLV) 1 and 2 antibodies, venereal diesease research laboratory (VDRL) test, hepatitis B antigen (HBsAg), Anti HCV (hepatitis C virus) antibody and antinuclear antibody were also negative. A low CD4+ T-cell count of 186 cells/mm 3 was the only abnormal finding. This can be a transient, non-specific observation in an infection and he was treated with appropriate antibiotics. [2]
Patient was re-evaluated after 3 months and his skin lesions had all subsided, but the CD4 count was still low at 180 cells/mm 3 . A repeat clinical and laboratory work up failed to detect any cause for the persistent lymphocytopenia. Hence, we arrived at the final diagnosis of ICL. [3] As the cluster of differentiation (CD) 4 count was below 200, he was prescribed cotrimoxazole prophylaxis and was urged to be under regular follow-up.
He returned 1 year later with pruritic, infiltrated papules, and plaques all over the body and significantly enlarged cervical, axillary, and inguinal lymph nodes [Figure - 1]a. CD4 count was 178 cells/mm 3 . Again there was no evidence of retroviral infection. Ultrasound abdomen revealed hepatosplenomegaly and retroperitoneal lymphadenopathy. Biopsy from an enlarged cervical node showed loss of normal architecture with moderate to marked infiltrate of atypical lymphocytes. Histology [Figure - 1]b of the skin lesions revealed dermal infiltrate of atypical lymphocytes without epidermotropism. On immunohistochemistry, the tumour cells were CD3 +ve [Figure - 2]a, CD4 +ve [Figure - 2]b, CD8+ve [Figure - 2]c and CD20-ve [Figure - 2]d pointing to a diagnosis of cutaneous T-cell NHL. The patient was referred to Hemato-Oncology Department, chemotherapy was initiated, but while on chemotherapy he developed septicemia and succumbed to death.
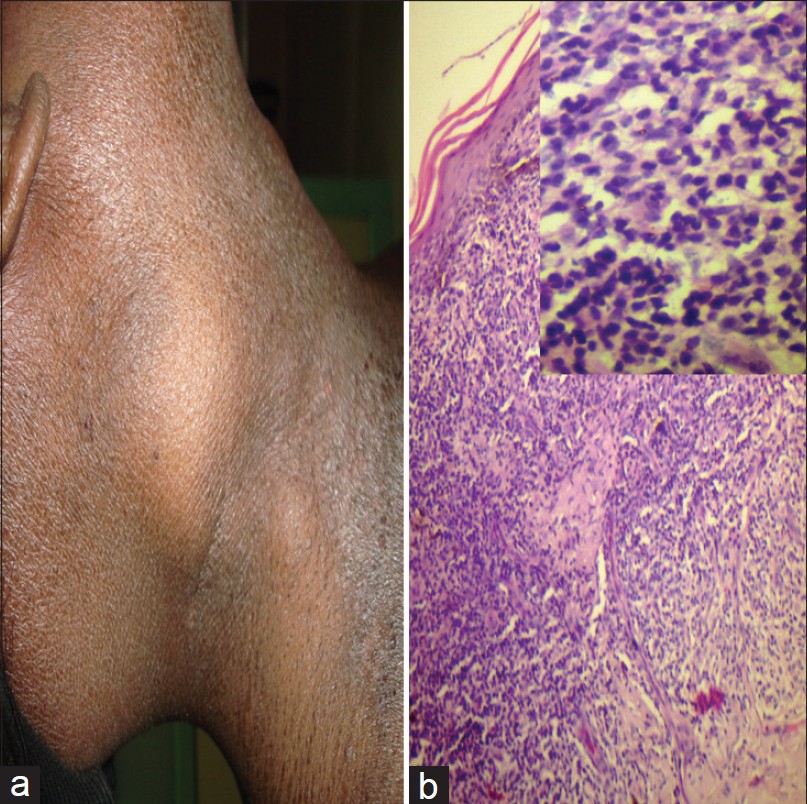 |
| Figure 1: (a) Enlarged cervical lymph node and infiltrated skin lesions. (b) Skin biopsy from the infiltrated plaque showing dermal infiltrate of atypical cells without epidermotropism (H and E, ×100). Inset: High power view of the same showing dermal infiltrate composed of atypical cells with irregular outline, scanty cytoplasm and hyperchromatic nuclei (H and E, ×400) |
 |
| Figure 2: (a) Dermal infiltrate showing CD3 positivity (Immunohistochemistry, ×400). (b) Dermal infiltrate showing CD4 positivity (Immunohistochemistry, ×400). (c) Dermal infiltrate showing CD8 positivity (Immunohistochemistry, ×100). (d) Dermal infiltrate showing CD20 negativity (Immunohistochemistry, ×100) |
CDC defined ICL in 1992 as (1) CD4+ T-lymphocyte level <300 cells/mm 3 or <20% of total lymphocytes, at a minimum of two separate time points, at least 6 weeks apart. (2) No serological evidence of HIV infection. (3) Absence of any defined immunodeficiency or therapy that can depress CD4 count. [3]
Various theories put forth to explain the etiology of ICL include diminished production of T cell precursors, accelerated T-cell apoptosis, biochemical failure of the CD3-T cell receptor pathway, defective generation of tumour necrosis factor (TNF)-α and interferon (IFN)-γ and antibodies against CD4+ T cells. [3] Clinical picture varies from an asymptomatic laboratory abnormality to life threatening complications such as opportunistic infections and neoplasms. [1] It is documented in one series that, in about one fifth of patients, lymphocytopenia resolved within 3 years of diagnosis whereas in the majority, most serious complications developed during the first 2 years of diagnosis. [1] Our patient manifested T-cell NHL 1 year after the confirmation of ICL.
Previous reports point to a predominance of Burkitt′s and other B-cell lymphomas in ICL, but our patient developed T-cell NHL, which is a rare occurrence. [3],[4] A T-cell lymphoma in the setting of CD4+ lymphocytopenia is hard to explain. A pathogen specific, immune response driven, CD4+ T-cell activation and turn-over is proposed to be the root cause of lymphocytopenia in ICL. [1] This constant activation may precipitate the monoclonal expansion of atypical cells in the remaining T-cell population. On searching the literature, we came across only one more instance of T-cell neoplasm in ICL and this was reported by Moradi et al. (angiocentric nasal T-cell lymphoma) in a 13-year-old boy. [5]
Current recommendations (including cotrimoxazole prophylaxis) for the treatment of ICL are based mainly on the experience with HIV patients. [1] IFN-γ has been tried to boost the depressed CD4 levels. [3]
We report this case to stress the need to consider ICL, in any patient (with no known immunodeficiency) presenting with opportunistic infections. Had it not been diagnosed earlier, lymphocytopenia in our patient would have been attributed to the lymphoma. Meticulous documentation and analysis of the clinical features and disease progression in those affected, may help us to improve the diagnostic definition and treatment options for this rare condition.
Acknowledgments
We express our sincere gratitude to Dr. Sunitha Balakrishnan, Asst. Professor, Department of Pathology for the help in analysing the histopathology specimens.
| 1. |
Zonios DI, Falloon J, Bennett JE, Shaw PA, Chaitt D, Baseler MW, et al. Idiopathic CD4+ lymphocytopenia: Natural history and prognostic factors. Blood 2008;112:287-94.
[Google Scholar]
|
| 2. |
Laurence J. T-cell subsets in health, infectious disease, and idiopathic CD4+ T lymphocytopenia. Ann Intern Med 1993;119:55-62.
[Google Scholar]
|
| 3. |
Luo L, Li T. Idiopathic CD4 lymphocytopenia and opportunistic infection - An update. FEMS Immunol Med Microbiol 2008;54:283-9.
[Google Scholar]
|
| 4. |
Hanamura I, Wakita A, Harada S, Tsuboi K, Komatsu H, Banno S, et al. Idiopathic CD4+ T-lymphocytopenia in a non-Hodgkin's lymphoma patient. Intern Med 1997;36:643-6.
[Google Scholar]
|
| 5. |
Moradi S, Chavoshzadeh Z, Izadyar M, Mahjoub F, Rezaei N. Angiocentric nasal T-cell lymphoma in a patient withidiopathic CD4+ lymphocytopenia. Iran J Allergy Asthma Immunol 2009;8:215-8.
[Google Scholar]
|
Fulltext Views
1,935
PDF downloads
1,725





![[Figure - 1]](#fig_ijdvl_2013_79_6_831_120748_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_6_831_120748_f2.jpg){kind=link}