Translate this page into:
A case of atypical scleromyxedema without gammopathy treated with cyclosporine
2 Department of Dermatology, �orlu Military Hospital, Tekirdag, Turkey
3 Department of Pathology, G�lhane School of Medicine, Ankara, Turkey
Correspondence Address:
G�rol A�ikg�z
Department of Dermatology, G�lhane School of Medicine, 06018 Etlik, Ankara
Turkey
| How to cite this article: A�ikg�z G, �zmen I, H�seynov S, Gamsizkan M, �aliskan E, Arca E, Ko� E. A case of atypical scleromyxedema without gammopathy treated with cyclosporine. Indian J Dermatol Venereol Leprol 2014;80:278 |
Sir,
Lichen myxedematosus is a disorder characterized by the formation of numerous lichenoid papules causing extensive thickening of the skin. Scleromyxedema, which is characterized by generalized lichenoid papules or scleroderma-like lesions, is considered to be generalized lichen myxedematosus or representative of the papular mucinosis group of cutaneous disorders. [1] The fibroblasts are triggered by an unknown mechanism resulting in excessive mucin deposition in dermis. The disease primarily affects the skin but extracutaneous manifestations including myositis, monoclonal gammopathy and neurological, cardiovascular, and renal involvement are well recognized. [2],[3] Herein, we describe a patient presenting with dermatological features of scleromyxedema and myositis without any monoclonal gammopathy.
A 23-year-old Caucasian man was referred to the outpatient clinic with a history of edema, diffuse skin thickening, and eruptions. In 1 month, the lesions had gradually increased and spread over the affected areas. His medical history and family history were unremarkable. Dermatological examination revealed diffuse erythema, induration and coarsening of the face (leonine facies), and diffuse widespread, erythematous, symmetric, waxy, shiny, small papules and linear plaques on face, forehead, neck, upper and lower extremities, and back [Figure - 1]. The other clinical findings were constricted mouth opening, muscle weakness of the upper and lower extremities, and sclerodactyly.
 |
| Figure 1: The patient before and after the oral cyclosporine therapy |
Skin biopsy revealed fibrosis, fibroblast proliferation [Figure - 2] and deposition of large amounts of basophilic dermal mucin [Figure - 3] in the papillary and upper reticular dermis. There was no vacuolar interface change in the basal layer of the epidermis.
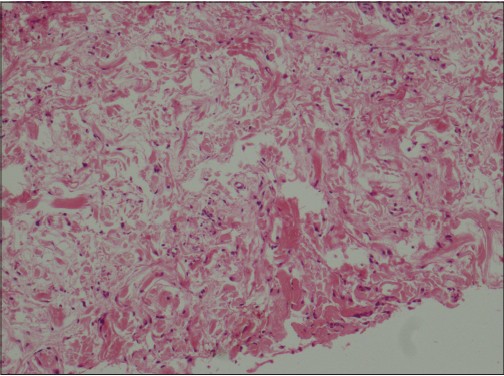 |
| Figure 2: Fibroblastic activity in reticular dermis (H and E, �200) |
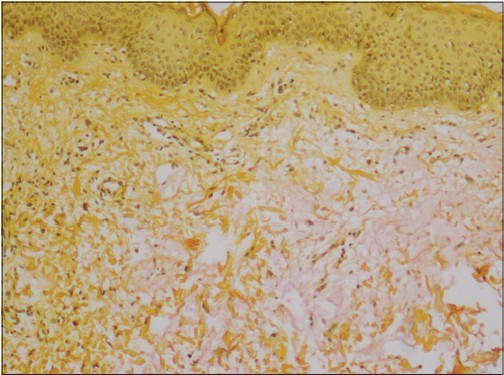 |
| Figure 3: Dermal mucin deposition seen on the right side of the image (Mucicarmine stain, �100) |
The serum lactate dehydrogenase level was elevated (724 U/L [NR: 220-450 U/L]). Other laboratory investigations including complete blood count and routine blood analysis including other muscle enzymes, thyroid function tests, serum protein electrophoresis, and serum immunoglobulin levels were all within normal limits. Antinuclear antibody, anti-dsDNA, anti-SSA, anti-SSB, anti-Sm, and anti-Scl-70 antibodies were also negative. Chest radiography and electrocardiography were normal with minimal mitral and tricuspid regurgitation detected on echocardiography. Deltoid muscle electromyography was performed because of muscle weakness and it revealed findings consistent with acute myositis. As dermatomyositis has occasionally been reported in association with lichen myxedematosus, we looked for evidence of the diagnosis but excluded it as there was no cutaneous and laboratory evidence.
The patient was diagnosed with atypical scleromyxedema and treatment with oral fluocortolone 60 mg daily was initiated. After 2 months, the dose was tapered to 40 mg/day. No clinical improvement was observed at the end of 3 months of steroid therapy except minimal recovery in muscle weakness. The treatment was switched to cyclosporine, 300 mg/day. Clinical findings regressed at the end of the first month and cyclosporine was tapered to 200 mg/day. To date, he is still being monitored and has minimal clinical findings including several persisting shiny papules on his back and shoulders [Figure - 1].
Lichen myxedematosus has been considered in three main clinico-pathological subsets: generalized papular and sclerodermoid form (scleromyxoedema), localized papular form, and atypical or intermediate form. [2] Diagnosis of scleromyxedema is based on the presence of: (i) a generalized papular and sclerodermoid eruption, (ii) mucin deposition, fibroblast proliferation and fibrosis on biopsy, and (iii) monoclonal gammopathy and absence of a thyroid disorder. The third atypical or intermediate form of lichen myxedematosus is described for patients with the generalized eruption who do not fulfill the diagnostic criteria of scleromyxedema. [2] The present patient demonstrated all diagnostic criteria of scleromyxedema except gammopathy, and therefore the diagnosis was atypical-intermediate form.
The pathogenesis of the disease remains unclear but a monoclonal gammopathy with elevated λ light chain of IgG, which is observed in more than 80% of patients, is considered a trigger factor for fibroblast proliferation and mucin production. [3] Although dermal mucin deposition is characteristic for scleromyxedema, it is not consistently found in other organs which are involved.
Numerous extracutaneous features and systemic involvement have been reported in the literature. Symptoms of myopathy which vary from myalgia to profound weakness are observed in 27% of patients. [4] Our patient complained of muscle weakness and acute myositis was demonstrated as a co-existing condition.
Treatment of scleromyxedema is very difficult and disappointing. Steroids, chemotherapeutic agents, retinoids, plasmapheresis, IVIG, thalidomide, chloroquin, radiotherapy, and autologous stem cell transplantation have been used with variable results. Saigoh et al. reported a patient who was treated with oral steroids, D-penicillamine, psoralen-UVA and plasmapheresis with no improvement. Thereafter, they started cyclosporine, 50-100 mg/day and after 4 months there was at least 50% improvement. [5] In the present patient, the treatment plan was changed to cyclosporine and significant clinical improvement was seen within 2 months.
In conclusion, scleromyxedema is a rare mucin deposition disorder that may present in numerous forms. Though many different methods have been attempted, there is no standard therapy and the results of each modality are quite variable. Although all patients may not respond as well our patient did, cyclosporine can be considered as a treatment option alone or as an adjunct to other therapies in patients with refractory scleromyxedema.
Acknowledgment
This case was presented as a poster at the AAD Summer Meeting, New York, USA, 3-7 August 2011.
| 1. |
Serdar ZA, Altunay IK, Yasar SP, Erfan GT, Gunes P. Generalized papular and sclerodermoid eruption: Scleromyxedema. Indian J Dermatol Venereol Leprol 2010;76:592.
[Google Scholar]
|
| 2. |
De D, Parsad D, Kanwar AJ, Saikia UN. Papules and sclerodermoid change in a middle-aged woman. Sceromyxoedema without gammopathy. Clin Exp Dermatol 2010;35:e54-5.
[Google Scholar]
|
| 3. |
Heymann WR. Scleromyxedema. J Am Acad Dermatol 2007;57:890-1.
[Google Scholar]
|
| 4. |
Tan E, Lau N, Yung A. Scleromyxoedema: A cause for unexplained encephalopathy and myositis. Clin Exp Dermatol 2010;35:746-8.
[Google Scholar]
|
| 5. |
Saigoh S, Tashiro A, Fujita S, Matsui M, Shibata S, Takeshita H, et al. Successful treatment of intractable scleromyxedema with cyclosporin A. Dermatology 2003;207:410-1.
[Google Scholar]
|
Fulltext Views
2,012
PDF downloads
1,221





![[Figure - 1]](#fig_ijdvl_2014_80_3_278_132272_u1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2014_80_3_278_132272_u2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2014_80_3_278_132272_u3.jpg){kind=link}