
Translate this page into:
A de novo case of ankyloblepharon, ectodermal defects, cleft lip/palate syndrome with TP63 mutation diagnosed prenatally
Corresponding author: Dr. Sirisha Varala, Department of Dermatology, Venereology, Leprosy, Osmania Medical College, Afzalgunj, Hyderabad, India. siru815@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Varala S, Challa S, Gugulothu N, Ananthula VK, Thakur RS. A de novo case of ankyloblepharon, ectodermal defects, cleft lip/palate syndrome with TP63 mutation diagnosed prenatally. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_152_2024
Dear Editor,
A 40-day-old infant, who was the first born to non-consanguineous parents, with the mode of delivery being emergency caesarean section at 35 weeks of gestation (appearance, pulse, grimace, activity, and respiration [APGAR] score of 7), presented with complete absence of hair on scalp and body, cleft lip and palate [Figure 1], anonychia of finger and toe nails, ankyloblepharon [Figure 2] and superficial erosions on scalp and body [Figure 3]. The skin was dry, shiny, and wrinkled all over the body. A routine antenatal scan in the mother revealed a cleft lip, following which whole exome sequencing was done on foetal amniotic fluid. A heterogeneous mutation in exon 13 (c.1655T>C) of tumour protein 63 (TP63) gene was detected. The pregnancy was however continued at the will of the parents and the child was born with classical features of ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. The child was treated with intravenous broad-spectrum antibiotics in the neonatal intensive care unit in view of sepsis. Erosions on the skin healed with post-inflammatory hypopigmentation. Eyelid adhesions lysed spontaneously aiding in the opening of the eyes. Surgery for cleft lip and palate was planned at a later date.
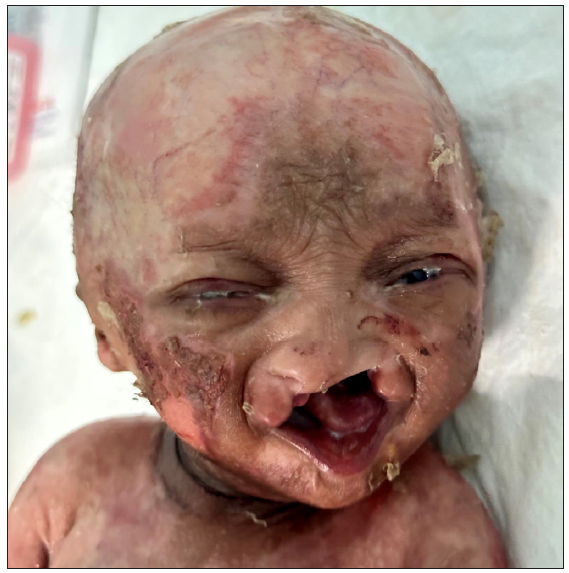
- Cleft lip.
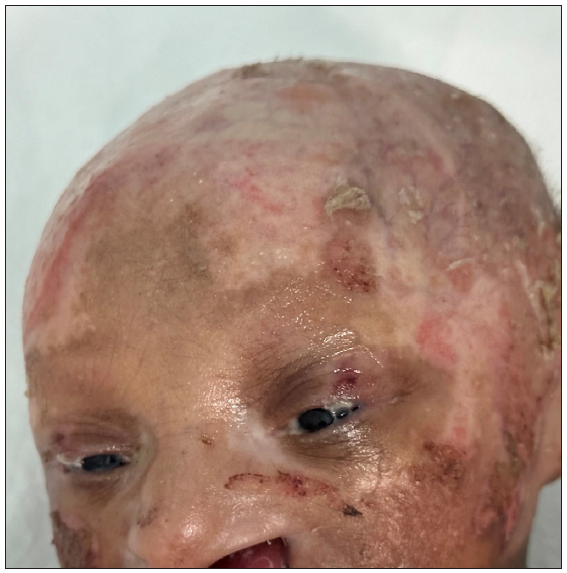
- Ankyloblepharon filiforme adnatum.
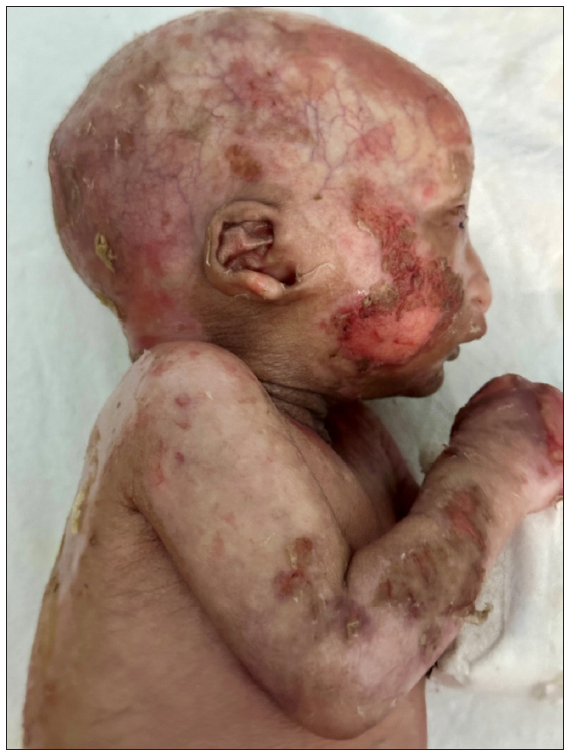
- Multiple erosions with crusting on scalp, face, and upper limb.
Ectodermal dysplasias are syndromes associated with abnormal development of skin, teeth, hair, and nails. The TP63, which is a transcription factor gene, has been linked with the development of ectodermal dysplasias which are characterized by orofacial clefting and limb abnormalities.1 These include six overlapping phenotypes:
-
Ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome (Online mendelian inheritance in man [OMIM] #106260) (which includes Rapp-Hodgkin [RH] syndrome [OMIM#129400])
-
Acro-dermo-ungual-lacrimal-tooth (ADULT) syndrome (OMIM#103285)
-
Ectrodactyly, ectodermal dysplasia, cleft lip/palate syndrome 3 (EEC3) (OMIM#604292)
-
Limb-mammary syndrome (OMIM#603543)
-
Split-hand/foot malformation type 4 (SHFM4) (OMIM#605289)
-
Isolated cleft lip/cleft palate (orofacial cleft 8) (OMIM#618149)
AEC syndrome was first described in 1976, by Hay and Wells.2 Hence, it is also known as Hay-Wells syndrome. A heterozygous mutation in the TP63 gene located on chromosome 3q28 is causative.3 Clinical features usually manifest at birth and are characterised by ectodermal defects, skin erosions, ankyloblepharon filiforme adnatum, cleft lip, and palate. Our patient had classical features of this triad, thereby fitting phenotypically into the diagnosis of AEC syndrome which was confirmed by prenatal genetic testing.
Ankyloblepharon filiforme adnatum is found in only 44% of cases, although it is regarded as a pathognomonic finding of AEC.4 Ankyloblepharon can manifest as complete fusion of the eyelids or partial fusion or even reduced palpebral fissure length. An erythrodermic presentation with multiple superficial erosions leading to potentially fatal infections with skin failure is quite common at birth.3 Scalp is a common site for erosions, leading to scarring alopecia. The erosions on the scalp in our case showed little tendency towards healing while those on the trunk and extremities healed with post-inflammatory hypopigmentation. Ninety-four per cent of the cases have hair defects like universal alopecia or coarse wiry hair, while more than 80% of patients show teeth and nail anomalies.4
Historically, TP63 syndromes have been considered as separate entities; however, various overlapping features have been found. Chiu et al. have described an infant with overlapping features of EEC and AEC and opined that TP63 disorders are allelic conditions with variable expression of the same spectrum of disease.1 Similarly, recent genetic molecular studies have also suggested that mutations in TP63 gene can present with a wide phenotypic spectrum, including AEC, EEC and RH syndrome.5
Genetic testing helps in the confirmation of diagnosis. In our case, a heterozygous missense variant in exon 13 of the TP63 gene involving the sterile alpha motif (SAM) domain with substitution of Serine for Phenylalanine at codon 552 (p.Phe552Ser) was detected. Targeted mutation analysis for the same in parents showed the absence of the variation, suggesting its de novo status. The initial American College of Medical Genetics and Genomics (ACMG) classification of this variant was ‘variant of unknown significance”. However, after confirming the de novo status, it was reclassified as ‘likely pathogenic’. The variant has been reported as ‘probably damaging’ by PolyPhen-2 and ‘damaging’ by sorting intolerant from tolerant (SIFT), likelihood ratio test (LRT), and Mutation taster 2. Full-length TP63 gene comprises of six domains, and pathogenic variations in the SAM domain are responsible for most of the cases of AEC and RH syndromes.6 Hori et al. reported a case of AEC/RH syndrome-like ectodermal dysplasia with a novel nonsense TP63 variant in a short isoform-specific exon which they speculated to be responsible for milder skin manifestations as seen in their case, while mutations in SAM and transactivation-inhibitory domains lead to severe skin lesions.7
There is no intellectual impairment in these patients; however, quality of life is greatly affected due to associated multi-organ abnormalities.6 Multidisciplinary management therefore should be offered to improve the quality of life.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- A case of ankyloblepharon, ectodermal dysplasia, and cleft lip/palate syndrome with ectrodactyly: Are the p63 syndromes distinct after all? Pediatr Dermatol. 2011;28:15-9.
- [CrossRef] [PubMed] [Google Scholar]
- The syndrome of ankyloblepharon, ectodermal defects and cleft lip and palate: An autosomal dominant condition. Br J Dermatol. 1976;94:277-89.
- [CrossRef] [PubMed] [Google Scholar]
- Novel missense mutation of the TP63 gene in a newborn with Hay-Wells/ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome: Clinical report and follow-up. Ital J Pediatr. 2021;47:196.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Pattern of p63 mutations and their phenotypes – update. Am J Med Genet A. 2006;140:1396-406.
- [CrossRef] [PubMed] [Google Scholar]
- Rapp-hodgkin syndrome: A review of the aspects of hair and hair color. J Am Acad Dermatol. 2005;53:729-35.
- [CrossRef] [PubMed] [Google Scholar]
- TP63-related disorders: Two case reports and a brief review of the literature. Dermatol Online J. 2021;15:27.
- [CrossRef] [PubMed] [Google Scholar]
- A novel TP63 variant in a patient with ankyloblepharon-ectodermal defect-cleft lip/palate syndrome and Rapp-Hodgkin syndrome-like ectodermal dysplasia. Hum Genome Var. 2022;9:17.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




