Translate this page into:
A new presentation of isolated cutaneous Rosai-Dorfman disease: Eruptive xanthoma-like lesions
2 Department of Dermatology, Andrology and STDs, Port Said University, Port Said, Egypt
Correspondence Address:
Mohammed Fawzy El-Kamel
Department of Dermatology, Andrology and STDs, Mansoura University, Mansoura, 35516
Egypt
| How to cite this article: El-Kamel MF, Selim MK, Gawad MM. A new presentation of isolated cutaneous Rosai-Dorfman disease: Eruptive xanthoma-like lesions. Indian J Dermatol Venereol Leprol 2020;86:158-161 |
Abstract
Rosai-Dorfman disease or sinus histiocytosis with massive lymphadenopathy is a benign lympho-histiocytic proliferative disorder initially described with bilateral painless lymphadenopathy (90 %), fever, leukocytosis, elevated ESR, anemia, and polyclonal hypergammaglobulinemia (90 %). Extranodal forms occur in 43% of cases, the skin being the most common site. Around 10% of patients have skin lesions and in 3%, the disease is limited exclusively to the skin. Here, we report a male patient who presented with pure cutaneous lesions which mimic eruptive xanthoma clinically. However, the diagnosis was established histo pathologically. So, high level of clinical suspension is critical to avoid missing such cases.
Introduction
Rosai-Dorfman disease is a non-Langerhans cell histiocytosis first described in 1965 by Destombes and recognized as a distinct clinicopathological entity by Rosai and Dorfman in 1969.[1],[2] Rosai-Dorfman disease presents with cervical adenopathy, most commonly in children or young adults (median age, 20 years), in those of African ancestry and male sex.[1] Extranodal disease occurs in 25-40%, is often widespread, involving skin, respiratory tract, soft tissue, paranasal sinuses, visceral organs, bone, central nervous system, genitourinary tract, and orbit.[2],[3] Around 10% of patients have skin lesions and in 3%, the disease is limited exclusively to the skin.[3] Cutaneous Rosai-Dorfman disease presents as solitary or numerous papules, nodules, plaques or as a combination of these.[4]
Case Report
A 46-year-old male reported appearance of asymptomatic widespread skin lesions 4 months ago. The lesions grew rapidly on the trunk and left thigh and later spread to involve other extremities and face within a span of 2 months. The patient had no related medical history and was otherwise healthy. The patient reported no systemic symptoms such as fever, malaise or weight loss. There was no organomegaly or lymph node enlargement.
Cutaneous examination revealed innumerable generalized papules mainly affecting the face and limbs. The lesions were yellowish to orange papules with confluence forming non infiltrated plaques mainly on the thighs, cheeks, temples and peri-auricular area [Figure - 1]. There was no mucosal or nail involvement. Our differential diagnoses were eruptive xanthoma, fibrofolliculoma, trichodiscomas, eruptive villous hair cysts as well as non-Langerhans cell histiocytosis. Routine laboratory tests, serum thyroid stimulating hormone, X-ray of the chest and long bones and abdominal ultrasonography were normal. Computed tomography scan was carried out from the level of the craniocervical junction down to the groin and was normal. Lipid profile and serum protein electrophoresis tests were normal. Eye examination was unremarkable.
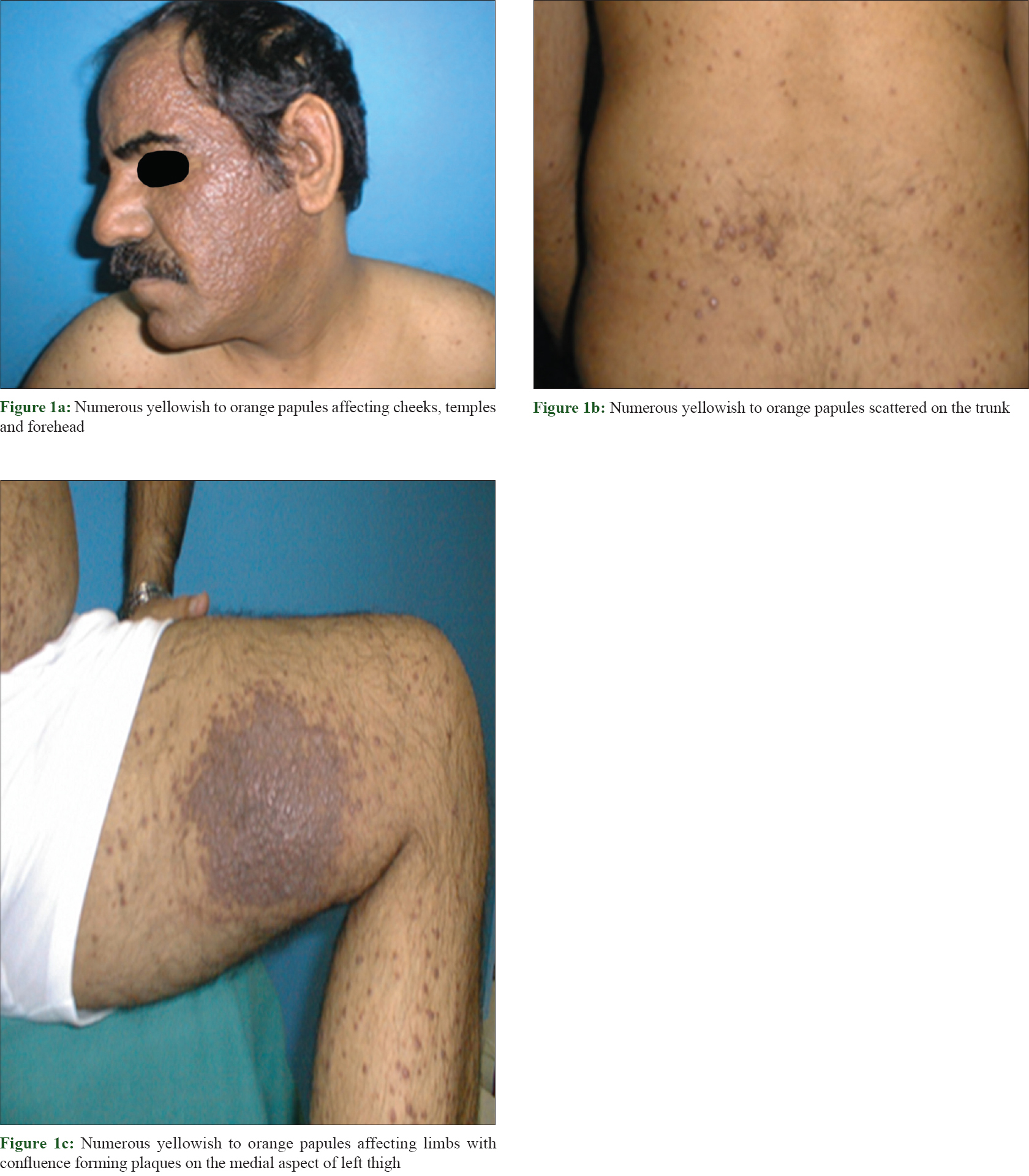 |
| Figure 1: |
A skin biopsy showed dermal infiltrate composed mainly of pale-staining macrophages with large cytoplasm and vesicular nuclei with evident nucleoli. Emperipolesis was evident. Lymphocytes, neutrophils and plasma cells were also found [Figure - 2]. Special stains including Giemsa stain, Fite Stain, Gram stain and periodic acid–Schiff were all negative. Immunohistochemistry revealed diffuse S-100 protein expression in the histiocyte cells, as well as positivity for CD68 and negativity for CD1a [Figure - 3].
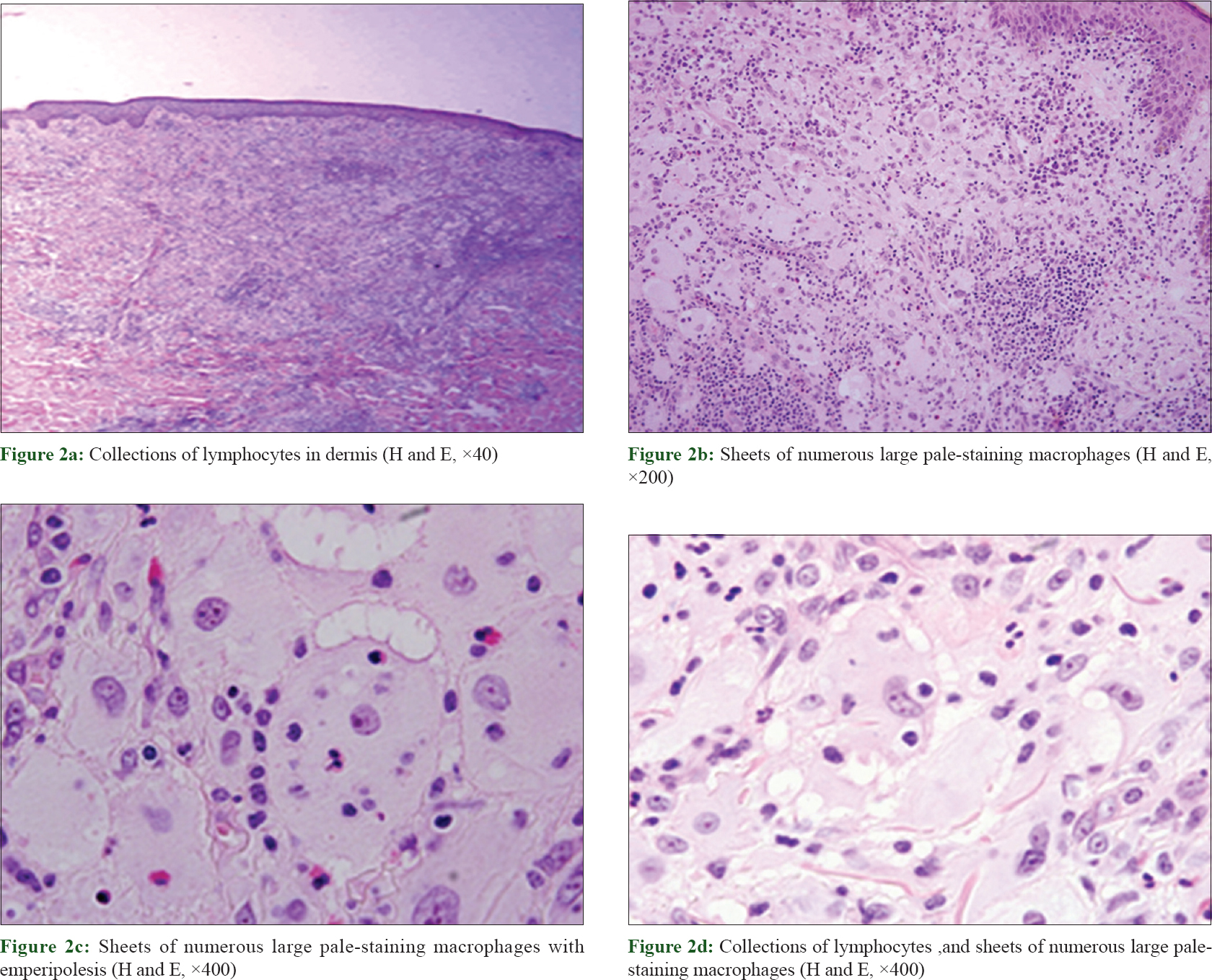 |
| Figure 2: |
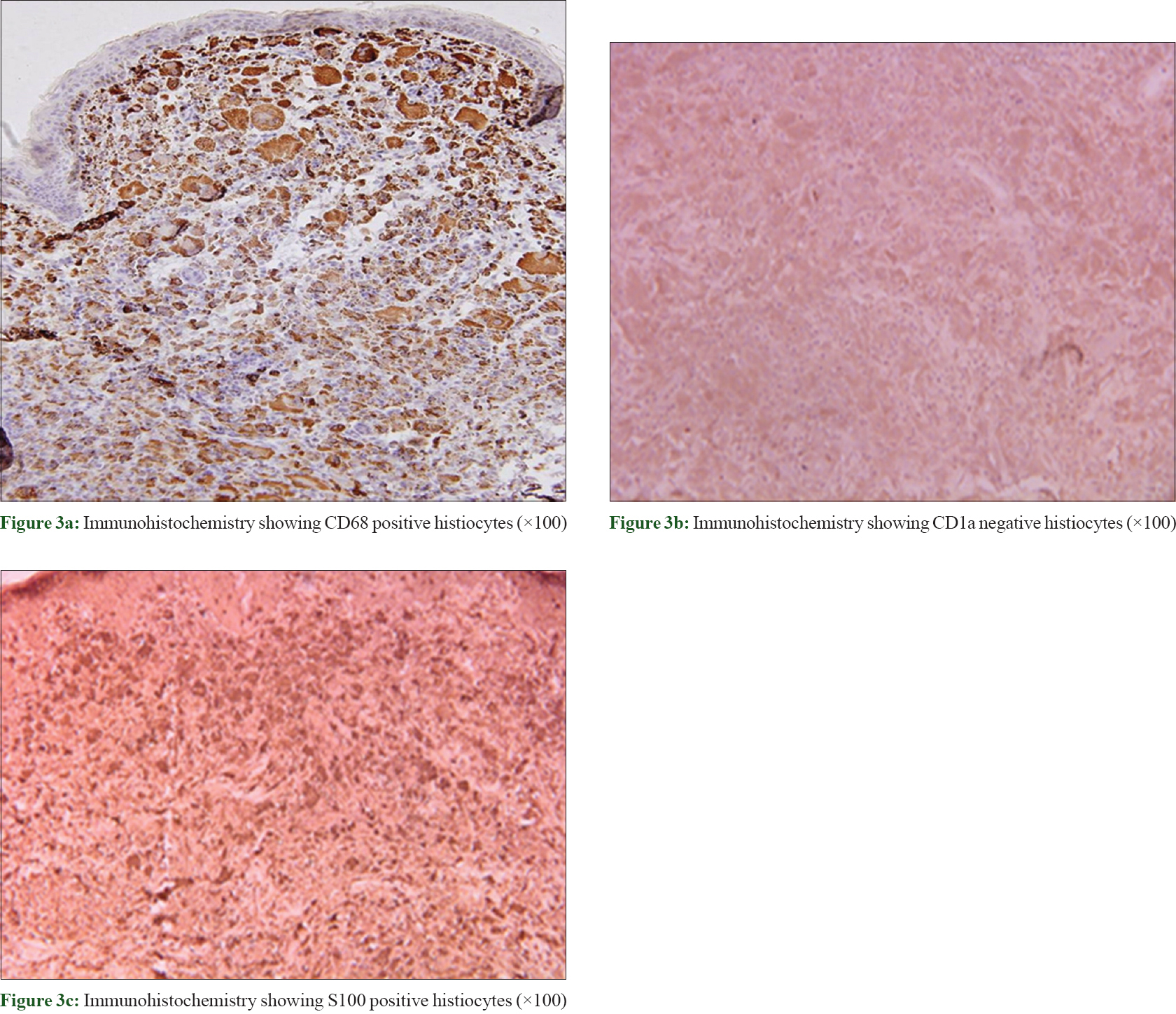 |
| Figure 3: |
Correlating the clinical and histopathological data, the diagnosis of exclusively cutaneous Rosai–Dorfman disease was established. Follow-up of the patient up to 2 years after his initial presentation showed that the disease remained stationary without any clinical, laboratory or radiological progression to the systemic classic form.
Discussion
The term cutaneous Rosai–Dorfman disease is used when involvement is limited only to the skin.[1],[2],[3] Approximately 10% of Rosai–Dorfman disease patients have skin lesions, and in 3%, the disease exclusively affects the skin.[4],[5] Cutaneous Rosai–Dorfman disease can present with papules, plaques, nodules and/or tumors of a brownish- or yellowish-erythematous color, varying in size from less than 1 cm to 30 cm, either localized or disseminated. The sites most commonly affected are trunk, head, neck, lower and upper extremities.[3] The lesions occasionally resemble psoriasis or acne.[6] We were unable to find any previous reports of cutaneous Rosai–Dorfman disease presenting as eruptive xanthoma like lesions in the literature.
The etiology of Rosai–Dorfman disease remains unknown. There are some reports that link the systemic form of the disease with herpes virus hominis-6 and 8 and Epstein–Barr virus. Researchers have also attempted to demonstrate this coexistence in a small series of cases in cutaneous Rosai–Dorfman disease; however, the results were conflicting.[3],[7] The finding of polyclonal gammopathy supports the hypothesis of a reactive process against infectious one.[1]
Histological findings in cutaneous Rosai–Dorfman disease are usually similar to those in the classic Rosai–Dorfman disease form. Typically, the dermis shows a diffuse infiltrate of histiocytes accompanied by a background infiltrate of lymphocytes and plasma cells. Emperipolesis, which represents intact lymphocytes within histiocytes, is common in cutaneous Rosai–Dorfman disease. Less often, the cytoplasm may contain plasma cells, neutrophils and red blood cells. Cutaneous Rosai–Dorfman disease histiocytes stain positively for S100 protein and CD68 but negatively for CD1a.[8]
Here, we report cutaneous Rosai–Dorfman disease manifesting in a new presentation – eruptive xanthoma-like eruption. The diagnosis of isolated cutaneous Rosai–Dorfman disease was made after excluding systemic involvement, either on first presentation or within 2 years of follow-up. Regarding treatment, we preferred wait and see policy, depending on the benign nature of the disease. Thus, cutaneous Rosai–Dorfman disease can be mistaken for a plethora of skin diseases, and can be included in the expanding list of skin imitators.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Pitamber HV, Grayson W. Five cases of cutaneous Rosai-Dorfman disease. Clin Exp Dermatol 2003;28:17-21.
[Google Scholar]
|
| 2. |
Salim A, Williamson M, Barker F, Hughes J. Steroid responsive cutaneous Rosai-Dorfman disease associated with uveitis and hypothyroidism. Clin Exp Dermatol 2002;27:277-9.
[Google Scholar]
|
| 3. |
Frater JL, Maddox JS, Obadiah JM, Hurley MY. Cutaneous Rosai-Dorfman disease: Comprehensive review of cases reported in the medical literature since 1990 and presentation of an illustrative case. J Cutan Med Surg 2006;10:281-90.
[Google Scholar]
|
| 4. |
Luz FB, Gaspar AP, Kalil-Gaspar N, Ramos-e-Silva M. Histiocytes and non-Langerhans cell histiocytoses in dermatology. An Bras Dermatol 2003;78:99-118.
[Google Scholar]
|
| 5. |
Van Zander J. Cutaneous Rosai-Dorfman disease. Dermatol Online J 2004;10:12.
[Google Scholar]
|
| 6. |
Uniyal SK, Beena KR, Ramesh V, Mukherjee A. Cutaneous Rosai-Dorfman disease preceding inguinal lymphadenopathy. Int J Dermatol 2002;41:404-6.
[Google Scholar]
|
| 7. |
Ortonne N, Fillet AM, Kosuge H, Bagot M, Frances C, Wechsler J, et al. Cutaneous destombes-Rosai-Dorfman disease: Absence of detection of HHV-6 and HHV-8 in skin. J Cutan Pathol 2002;29:113-8.
[Google Scholar]
|
| 8. |
Weitzman S, Jaffe R. Uncommon histiocytic disorders: The non-Langerhans cell histiocytoses. Pediatr Blood Cancer 2005;45:256-64.
[Google Scholar]
|
Fulltext Views
5,162
PDF downloads
3,328





![[Figure - 1]](#fig_ijdvl_2020_86_2_158_243619_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_2_158_243619_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2020_86_2_158_243619_f3.jpg){kind=link}