Translate this page into:
A noncancerous variant of xeroderma pigmentosum type D associated with novel heterozygous missense ERCC2 gene mutation
2 Department of Obstretics and Gynecology, Dr. S.N. Medical College, AIIMS Jodhpur, Rajasthan, India
3 Department of Medicine, Dr. S.N. Medical College, AIIMS Jodhpur, Rajasthan, India
Correspondence Address:
Shyama Choudhary
5, Ramnagar, Behind RTO Office, Jodhpur - 342 010, Rajasthan
India
| How to cite this article: Choudhary S, Parakh M, Suthar K, Parakh P, Khichar S. A noncancerous variant of xeroderma pigmentosum type D associated with novel heterozygous missense ERCC2 gene mutation. Indian J Dermatol Venereol Leprol 2017;83:594-595 |
Sir,
Xeroderma pigmentosum is an autosomal recessive genetic disease caused by nucleotide excision repair defect leading to a defective repair of DNA damaged by ultraviolet radiation.[1] Worldwide prevalence in all races is 1–4% per million. The disease begins around the age of 1–2 years with photosensitivity and burning sensation after nominal sun exposure in 60% of cases causing hyperpigmentation and ichthyosis in sun exposed areas, a 1000-fold increase in the risk of basal cell carcinoma, squamous cell carcinoma and melanoma of skin and eyes.[2] The median age of first cutaneous neoplasm is under 10 years. Ocular abnormalities include photophobia, ectropion, conjunctival infection, keratitis and incidence of tumors like squamous cell carcinoma, melanoma and epithelioma. Progressive neurologic symptoms including cognitive impairment, acquired microcephaly, difficulty in swallowing, abnormal motor activity, areflexia, sensorineural hearing loss and abnormal speech are present in about 25% of affected patients.[3] We report three siblings with xeroderma pigmentosum with skin, ocular, and neurological manifestations.
Case 1: An 11-year-old male child presented with eczematous skin lesions since the age of 2 years on sun exposed parts, watering of eyes for 1 year and poor vision. He had appropriate neurodevelopment until 3–4 years of age and gradually developed dysarthria, cognitive impairment, behavioral abnormalities and crouching gait. On examination, he had pallor, short stature, bilaterally hazy cornea, lentigenes [Figure 1a], bilateral foot drop leading to pes cavus, cerebellar dysarthia and crouching gait. Nerve conduction study was suggestive of an axonal demyelinating (predominantly axonal) neuropathy of both lower extremities. His brainstem evoked response audiometry was normal. Skin biopsy was not done as there was no suspected premalignant or malignant lesion (actinic keratosis or early basal cell carcinoma/squamous cell carcinoma).
 |
| Figure 1a: Three siblings with short stature, bilaterally hazy cornea and lentigenes over skin |
Case 2: The 14-year-old elder sister of case-1 presented with similar clinical features.
Case3: Youngest sibling of case-1 was an 8-year-old female child who presented with skin lesions and diminution of vision since last 2.5 years but had no neurological abnormality yet.
Pedigree analysis is presented in [Figure 1b]. None of the three siblings had any skin or ocular carcinoma but two of them manifested with axonal neuropathies. Corneal opacity and skin lesions improved by avoiding sun exposure, using ultraviolet preventing goggles, and sunscreen lotions on follow up.
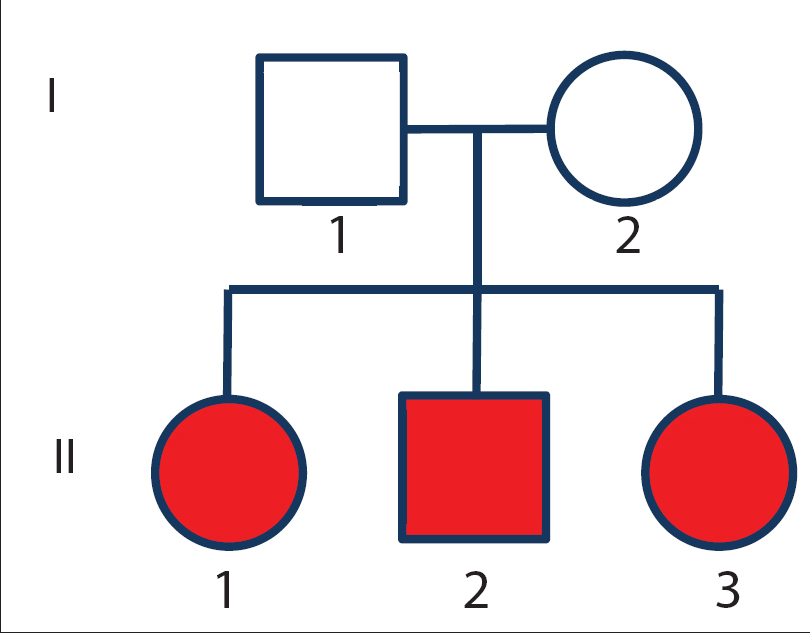 |
| Figure 1b: Pedigree of family |
On Target gene sequencing, two heterozygous variations in the ERCC2 gene were detected in all the three siblings [Figure 1c]. A heterozygous missense variation in exon 8 of the ERCC2 gene (chr19:45867700) that results in the amino acid substitution of Asparagine for Aspartic acid at codon 234 (p.D234N; ENST00000391945) and a heterozygous missense variation in exon 21 of the ERCC2 gene (chr19:45855768) that results in the amino acid substitution of Glycine for Aspartic acid at codon 681 (p.D681G; ENST00000391945) was detected. The parents were heterozygous for one variant each. Both variants were also extensively analyzed for pathogenicity and analysis revealed the variants are predicted to be probably damaging by PolyPhen-2 and damaging by SIFT, LRT and Mutation Taster.
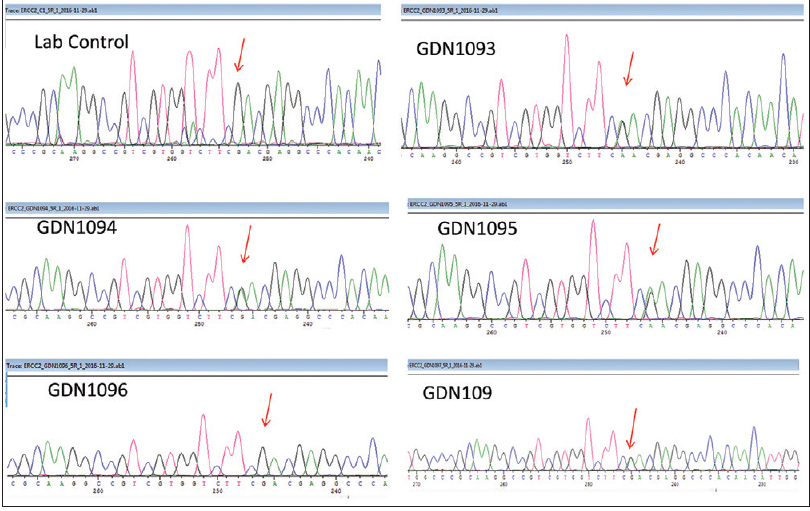 |
| Figure 1c: Targeted amplicon sequencing showing the compound heterozygous variation in all the affected siblings |
Absence of skin and eye cancer in our patients even at age of 14 years made us think whether a novel mutation in ERCC2 gene produces a noncancerous phenotype of XP. There is one case report from Japan of such mild presentation with other novel mutation in ERCC2.[4]
Mutations in the human XPD helicase gene are mainly single residue changes; yet they cause three strikingly different genetic disorders: XP, XP/CS (cockayne syndrome), and trichothiodystrophy (TTD). Although all three diseases share a photo-sensitivity phenotype, they differ greatly in their predispositions to cancer or accelerated aging. XP patients show several thousand-fold increase in skin cancer, whereas neither CS nor TTD patients shows an increase in the cancer incidence despite photosensitivity. Furthermore, both CS and TTD are premature aging diseases plus developmental disorders.[5] A previous paper reporting a patient with compound heterozygous variant with D681N suggests the variant might cause less severe manifestations of disease.[5]
In general, each mutational change in the XPD gene is specific for a particular clinical phenotype. The aforementioned case report significantly contributes to the understanding of genotype phenotype correlations in children with XP not having ocular or dermatologic malignancies. In this perspective, it is very important to establish a genetic diagnosis in children with XP to exactly prognosticate these patients and do an accurate genetic counseling of these families. A longer follow-up is required before conclusively saying that this is a noncancerous variant of XP and these families should be followed up life-long to better understand the natural history of this novel genotype.
Acknowledgments
Dr. Vinod Scaria (Scientist at CSIR-IGIB), and Dr. Mohammed Faruq and also CSIR Grant GOMED for performing Sanger sequencing of the family under guardian project.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. Nature 1968;218:652-6.
[Google Scholar]
|
| 2. |
Hasan S, Khan MA. Xeroderma pigmentosum with desquamative gingivitis a rare case report and detailed review of literature. J Cosmet Dermatol Sci Appl 2011;1:164-70.
[Google Scholar]
|
| 3. |
Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 1987;123:241-50.
[Google Scholar]
|
| 4. |
Ono R, Masaki T, Mayca Pozo F, Nakazawa Y, Swagemakers SM, Nakano E, et al. A10-year follow-up of a child with mild case of xeroderma pigmentosum complementation group D diagnosed by whole-genome sequencing. Photodermatol Photoimmunol Photomed 2016;32:174-80.
[Google Scholar]
|
| 5. |
Lehmann AR. The xeroderma pigmentosum group D (XPD) gene: One gene, two functions, three diseases. Genes Dev 2001;15:15-23.
[Google Scholar]
|
Fulltext Views
3,191
PDF downloads
1,310




