Translate this page into:
A novel H1 mutation in keratin 6a in an infant with pachyonychia congenita
2 Molecular Genetics Laboratory, Prenatal Diagnosis Center of Sichuan Province, West China Second University Hospital, Sichuan University, Chengdu, China
Correspondence Address:
Chaomin Wan
No. 17 Section Three, Ren Min Nan Lu Avenue, Chengdu, 610041, Sichuan
China
| How to cite this article: Luo S, Luo Q, Zhang H, Wan C. A novel H1 mutation in keratin 6a in an infant with pachyonychia congenita. Indian J Dermatol Venereol Leprol 2015;81:385-387 |
Abstract
Pachyonychia congenita (PC) is a rare genetic disorder which is inherited in an autosomal dominant pattern. We report a sporadic novel H1 mutation in the KRT6A gene (c. 428G>A/p.Ser143Asn) in a Chinese infant patient. The mutation is concurrent with a single-nucleotide polymorphism and resulted in a serine for asparagine substitution in H1 subdomain of KRT6A chain next to the rod domain. The infant showed the classic symptoms of pachyonychia congenita. Conclusion: The heterozygous missense mutation c. 428G > A/p.Ser143Asn in KRT6A exon 1 may cause severe disease.INTRODUCTION
Pachyonychia congenita (PC, OMIM 167200 and 167210) is a rare autosomal dominant genetic disease that mainly affects the nails, palmo-plantar skin and oral mucosa. The greatest impact on patients′ life is widely considered to be due to severe plantar pain secondary to thick keratoderma. [1] Based on the causative mutations identified in KRT6A, KRT6B, KRT6C, KRT16, and KRT17, pachyonychia congenita is subdivided into five subtypes namely PC-K6a, PC-K6b, PC-K6c, PC-K16, and PC-K17 respectively. [1],[2] The clinical phenotypes overlap substantially across PC-K6a, PC-K16, PC-K6b, and PC-K17; thick toenails and painful plantar keratoderma, are the major phenotypic features; other common clinical findings include thick fingernails, oral leukokeratosis, palmar keratoderma, follicular hyperkeratosis, cysts including epidermal inclusion cysts and pilosebaceous cysts, hoarseness, and natal teeth. [2],[3] Currently, information regarding clinical the phenotype of mutations in KRT6C is limited to a small number of reported cases. The individuals present with a milder phenotype than that caused by mutation in other genes, usually manifesting palmo-plantar keratoderma with only mild or no nail changes and plantar pain. [3],[4],[5],[6],[7] The age at onset and some phenotypic characteristics may help the clinician to suspect a specific subtype before genotyping is completed. [2],[3] Herein, we report a novel H1 mutation in KRT6A in a Chinese infant.
Case Report
A 3-month old female child presented with hoarseness, abnormal nails and oral mucosal plaques since birth. Nails appeared yellowish at birth and had gradually darkened and thickened subsequently [Figure - 1]a. At one week of age she was noticed to have a whitish plaque on the palatal mucosa which was diagnosed as "thrush" with associated "onychomycosis". Laboratory tests however, could not demonstrate fungus and she did not respond to itraconazole treatment. On examination, follicular hyperkeratosis was detected on both ears which was seen to progress to involve her back, buttocks and thighs by 1 year of age. At around 1 years of age, she was found to develop blisters on her soles after walking [Figure - 1]c. A detailed family history did not reveal any family member affected by a similar illness.
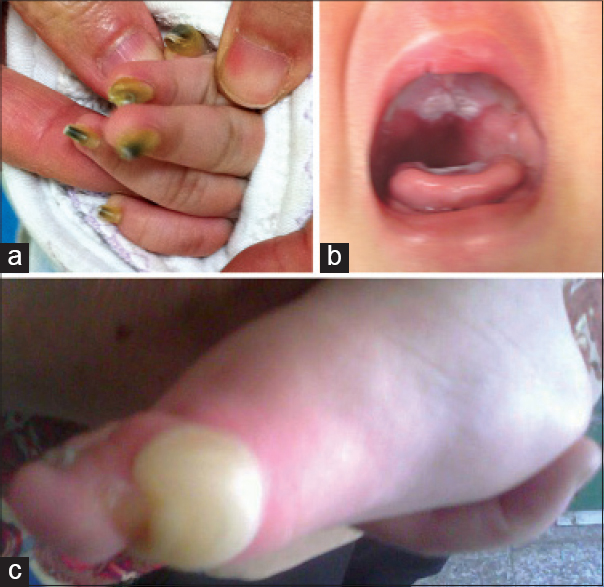 |
| Figure 1: Clinical features of the infant. (a) Hypertrophic nails at 3½ months of age (b) Oral leukokeratosis at the age of 6 months (c) Blisters on her feet at the age of 1½ years |
After informed consent was obtained, genomic deoxyribonucleic acid (gDNA) was extracted from whole blood samples taken from the patient and 12 of her family members [Figure - 2]. Polymerase chain reaction (PCR) and DNA sequence analysis of all exons in KRT6A, KRT16, KRT6B and KRT17 were performed by using Platinum® Taq DNA polymerase (Life technologies Invitrogen, USA), ABI Gene Amp PCR System 9700 PCR instrument (Life technologies Applied Biosystems, USA), and ABI 3730XL type DNA sequencer (Life technologies Applied Biosystems, USA). In addition, gDNA from 105 unrelated healthy Chinese individuals was used as controls.
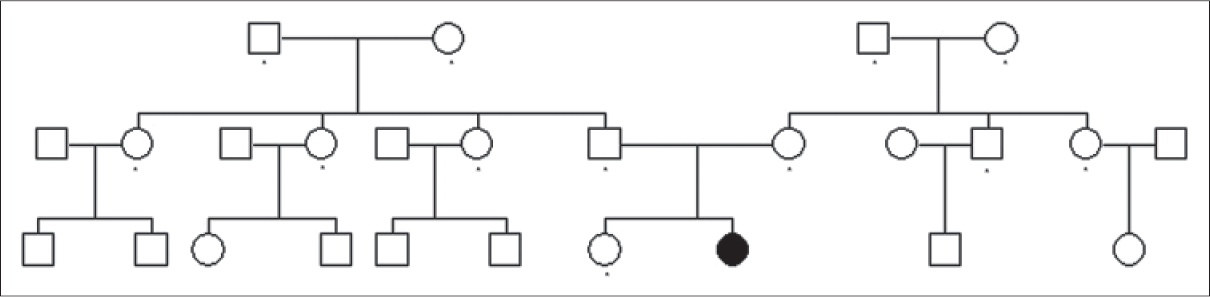 |
| Figure 2: Pedigree chart of the family of proband: Dots in the pedigree indicate family members who were examined and were found not to carry the mutation c.289G>A/p.Gly97Arg |
This study was approved by the Committee of Science and Ethics of the West China Second Hospital, Sichuan University.
Results and Discussion
This 2-year-old patient had thickened toenails and fingernails, plantar keratoderma, oral leukokeratosis, hoarseness and follicular hyperkeratosis without natal teeth or steatocystoma multiplex. Both the thickened toenails and plantar keratoderma are major clinical features of pachyonychia congenita across all mutation subtypes. [2] We were unable to ascertain the occurrence of plantar pain because she was too young to express her pain clearly. Concurrent toenail and fingernail thickening since birth, oral leukokeratosis, hoarseness and follicular hyperkeratosis are findings that occur more frequently in the PC-K6a subtype than in other subtypes. [3]
Amplification and DNA sequencing did not reveal any mutation in all KRT16, KRT6B, and KRT17 exons. However, one heterozygous missense mutation was found in KRT6A exon 1, c. 428G>A/p.Ser143Asn [Figure - 3]. This mutation was not detected in any of the 105 controls and her family members including her father (for whom DNA fingerprinting analysis was done to establish paternity). This suggests a pathogenic mutation rather than a rare single-nucleotide polymorphism (SNP). This mutation occurred in the H1 subdomain of the K6A protein just before the rod domain. Early studies indicate that the H1 subdomain is important in the alignment of the nearest neighboring molecules in the keratin intermediate filament structure. [8],[9] Mutations in the H1 domain do occur in other keratins, including KRT16 ; however, only two cases with mutation in H1 domain in KRT6A have been reported by Wilson et al. [1],[7]
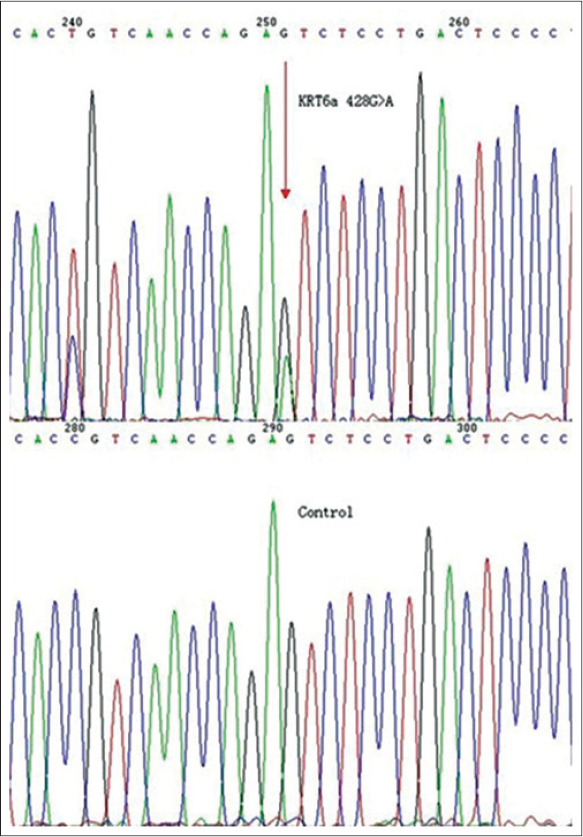 |
| Figure 3: Mutation analysis of KRT6A showing a heterozygous missense mutation KRT6A c.428G>A/p.Ser143Asn and the sequence of a control individual |
Our case in addition also demonstrated the nucleotide change c. 289G>A/p.Gly97Arg, which is a known single nucleotide polymorphism (SNP) in the dbSNP database (accession number: rs200254647). This nucleotide change was not detected in any of her family members.
ACKNOWLEDGMENTS
The authors express their gratitude to the patient and her family for their participation in the study.
| 1. |
Wilson NJ, Leachman SA, Hansen CD, McMullan AC, Milstone LM, Schwartz ME, et al. A large mutational study in pachyonychia congenita. J Invest Dermatol 2011;131:1018-24.
[Google Scholar]
|
| 2. |
Eliason MJ, Leachman SA, Feng BJ, Schwartz ME, Hansen CD. A review of the clinical phenotype of 254 patients with genetically confirmed pachyonychia congenita. J Am Acad Dermatol 2012;67:680-6.
[Google Scholar]
|
| 3. |
Shah S, Boen M, Kenner-Bell B, Schwartz M, Rademaker A, Paller AS. Pachyonychia Congenita in Pediatric Patients: Natural History, Features, and Impact. JAMA Dermatol 2013;150:146-53.
[Google Scholar]
|
| 4. |
Akasaka E, Nakano H, Nakano A, Toyomaki Y, Takiyoshi N, Rokunhe D, et al. Diffuse and focal palmoplantar keratoderma can bu caused by a keratin 6c mutation. Br J Dermatol 2011;165:1290-2.
[Google Scholar]
|
| 5. |
Kubo A, Oura Y, Hirano T, Aoyama Y, Sato S, Nakamura K, et al. Collapse of the keratin filament network through the expression of mutant keratin 6c observed in a case of focal plantar keratoderma. J Dermatol 2013;40:553-7.
[Google Scholar]
|
| 6. |
Wilson NJ, Messenger AG, Leachman SA, O′Toole EA, Lane EB, McLean WH, et al. Keratin K6c mutations cause focal palmoplantar keratoderma. J Invest Dermatol 2010;130:425-9.
[Google Scholar]
|
| 7. |
Wilson NJ, O′Toole E, Milstone LM, Hansen CD, Shepherd AA, AI-Asadi E, et al. The molecular genetic analysis of the expanding pachyonychia congenita case collection. Br J Dermatol 2014;171:345-55.
[Google Scholar]
|
| 8. |
Steinert PM. Structure, function, and dynamics of keratin intermediate filaments. J Invest Dermatol 1993;100:729-34.
[Google Scholar]
|
| 9. |
Steinert PM, Parry DA. The conserved H1 domain of the type II keratin 1 chain plays an essential role in the alignment of nearest neighbor molecules in mouse and human keratin 1/keratin 10 intermediate filaments at the two- to four-molecule level of structure. J Biol Chem 1993;268:2878-87.
[Google Scholar]
|
Fulltext Views
1,817
PDF downloads
1,557





![[Figure - 1]](#fig_ijdvl_2015_81_4_385_158651_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_4_385_158651_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_4_385_158651_f3.jpg){kind=link}