
Translate this page into:
A novel mutation in autoimmune polyglandular syndrome type 1 with erythematous facial papules
Corresponding author: Dr. Abhijit Saha, Department of Paediatric Dermatology, Institute of Child Health, Kolkata, India. drabhijit.saha812@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Saha A, Bashir S, Sahana MS, Dhar S. A novel mutation in autoimmune polyglandular syndrome type 1 with erythematous facial papules. Indian J Dermatol Venereol Leprol. 2025;91:248-50. doi: 10.25259/IJDVL_1388_2023
Dear Editor,
Autoimmune polyendocrine syndromes refer to a diverse group of clinical conditions characterised by loss of immune tolerance resulting in functional impairment of multiple endocrine glands.1 Autoimmune polyendocrine syndrome type 1 (APS-1) is a rare disorder resulting from autosomal recessive mutations of the autoimmune regulatory (AIRE) gene. This gene is a transcription factor primarily expressed in the thymus by medullary thymic epithelial cells as well as in secondary lymphoid organs. It facilitates the local transcription of organ-specific proteins that are normally produced in peripheral tissues. This process enables the negative selection of self-reactive T cells, preventing autoimmune responses.
In addition to the classical triad of chronic mucocutaneous candidiasis, hypoparathyroidism and adrenal insufficiency, APS-1 can have a myriad of endocrine and non-endocrine presentations. We present two siblings with APS-1 who had a similar clinical course and presentation of the syndrome. The gene sequencing, in addition to a known mutation, revealed a novel mutation in the AIRE gene that has not been previously reported.
A 9-year-old girl born of non-consanguineous parents presented with multiple, pruritic, erythematous, scaly, and crusted papules on the face, neck, chest, upper back, and axillae since 3 years of age [Figure 1]. Some of the lesions had healed with hypopigmentation after treatment with antifungals in the form of oral itraconazole 200 mg per day for 7 days and topical ketoconazole 2% cream twice daily for 4 weeks. [Figure 2]. Concomitant to the skin lesions the child had also developed recurrent whitish non-scrapable plaques with subsequent ulcerations in the oral mucosa, lips, and tongue, suggestive of chronic candidiasis of the oral cavity. Angular stomatitis was also noted at the time of presentation. The child had a history of abnormal posturing of both hands and a clear Trousseau’s sign could be elicited on physical examination. On dental examination, dental enamel hypoplasia was also noted [Figure 3]. No clinical or biochemical evidence of Addison’s disease was found. Serum calcium and parathyroid hormone levels revealed marked hypocalcemia and abnormally low parathyroid hormone levels. Parents of the child did not consent for a skin biopsy. Based on these findings a diagnosis of APS 1 was made. A clinical exome assay was carried out and targeted gene sequencing by polymerase chain reaction amplification of the protein coding regions of AIRE gene revealed a compound heterozygosity for c.1045C>T and c.967_979 del mutations in the AIRE gene. The younger brother of the child had cutaneous and oral lesions similar to the present patient and was also diagnosed with APS-1 [Figure 4]. Genetic testing in the younger sibling could not be carried out owing to the parents’ financial constraints.
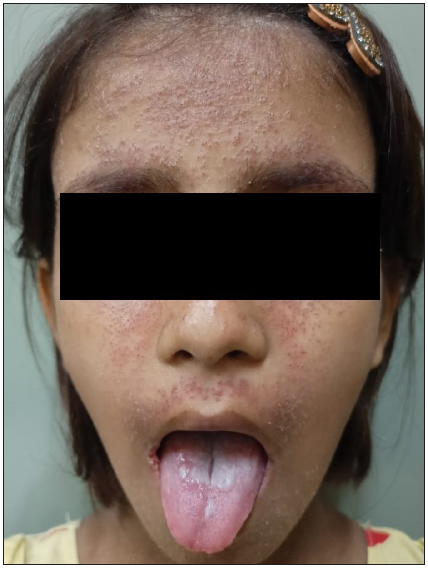
- Erythematous, scaly, and crusted papules on the face, neck, and chest with oral candidiasis.
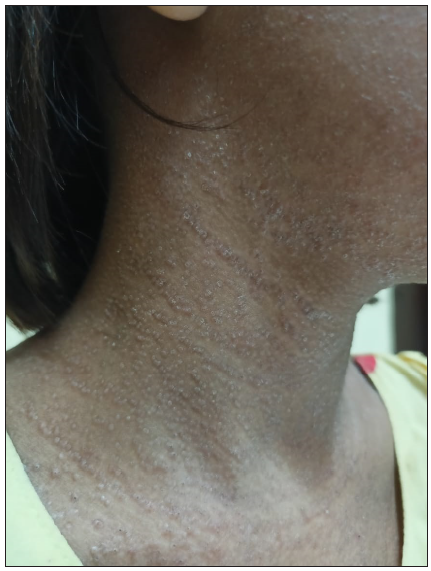
- Lesions treated with antifungals had healed with hypopigmentation at places.
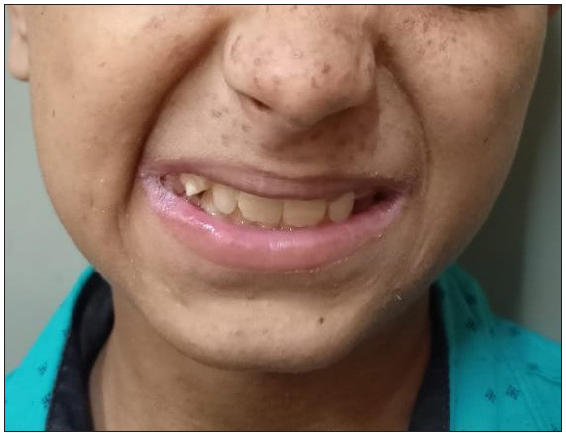
- Dental enamel hypoplasia.
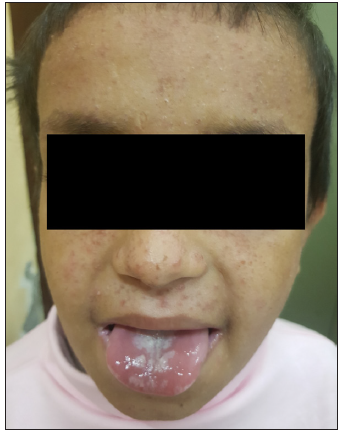
- The younger brother of the child with similar cutaneous lesions and oral candidiasis.
APS-1 is characterised by three cardinal components, i.e. chronic mucocutaneous candidiasis, hypoparathyroidism, and primary adrenal insufficiency (Addison’s disease). A definitive diagnosis requires the presence of two of the three components. In addition, APS-1 patients may develop several other endocrine and non-endocrine autoimmune disorders.2 Dental enamel hypoplasia is a commonly reported ectodermal disorder in these patients, while alopecia and vitiligo are less common. Both siblings in our report showed atypical cutaneous lesions on their face and upper bodies. Cutaneous lesions appear to be a rare manifestation of APS-1, among which urticaria and maculopapular or morbilliform rash are the most commonly reported.3 APS-1 patients have an increased mortality risk due to malignancies, adrenal and hypocalcemic crises. Certain conditions induced by aberrant autoimmune responses include hepatitis, nephritis, and pneumonitis.
APS-1 commonly results from biallelic AIRE mutations but large deletions or non-coding gene variants and non-AIRE gene variations have also been reported.4 A non-sense mutation in exon 6 (R257X) and a 13 base-pair (bp) deletion (967_979del13 bp) in exon 8 are the two most common mutations found in >95% of cases.5 While a common 13 bp deletion in exon 8 (967_979 del 13bp) was noted in the present patient, a novel heterogeneous non-sense mutation resulting from C to T point mutation at nucleotide 1045 in exon 9 of AIRE gene(c.1045C>T) was also detected and a premature truncation of the protein at codon 349 was predicted. At the protein level, this mutation is denoted as p.Gln349Ter. The p.Gln349Ter variant has not been reported in 1000 genomes databases and has a minor allele frequency of 0.0006% in the Genome Aggregation Database. By Mutation Taster 2, the in-silico prediction of the variant was found to be damaging. Based on these evidences, this novel AIRE variation was classified as a pathogenic variant.
The diagnosis of APS-1 needs a high index of suspicion. In patients with incomplete or unusual presentations, sequencing analysis of AIRE can be utilised as a useful screening method. Also, variations in genotype mutation as observed in the present patient, can affect the resultant phenotypes.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Autoimmune polyendocrine syndromes. N Engl J Med. 2018;378:1132-41.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Autoimmune polyendocrine syndrome type 1: An Italian survey on 158 patients. J Endocrinol Invest. 2021;44:2493-510.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J Clin Endocrinol Metab. 2006;91:2843-50.
- [CrossRef] [PubMed] [Google Scholar]
- Lessons from primary immunodeficiencies: Autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol Rev. 2019;287:103-20.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Positional cloning of the APECED gene. Nat Genet. 1997;17:393-8.
- [CrossRef] [PubMed] [Google Scholar]




