
Translate this page into:
A novel mutation in neurofibromatosis type 1
Corresponding author: Dr. Zaixing Wang, Department of Dermatology, First Affiliated Hospital of Anhui Medical University, Hefei, Anhui, China. wzx2370@163.com
-
Received: ,
Accepted: ,
How to cite this article: Luo Z, Tao X, Jin Z, Wang Z. A novel mutation in neurofibromatosis type 1. Indian J Dermatol Venereol Leprol 2023;89:453-5.
Dear Editor,
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder with a global prevalence of about 0.03% caused by mutations in the neurofibromatosis type 1 gene at 17q11.2, which encodes neurofibromin. Approximately half of the patients have family history and the others have sporadic mutations.1 Typical symptoms of neurofibromatosis type 1 include café-au-lait macules, cutaneous neurofibroma or plexiform neurofibroma, axillary or inguinal freckles, iris Lisch nodules, optic pathway gliomas, skeletal dysplasia, behavioural defects and cognitive impairment.
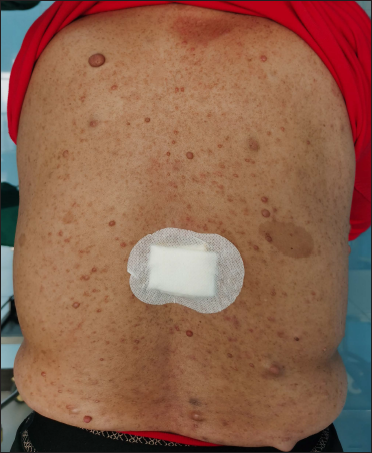
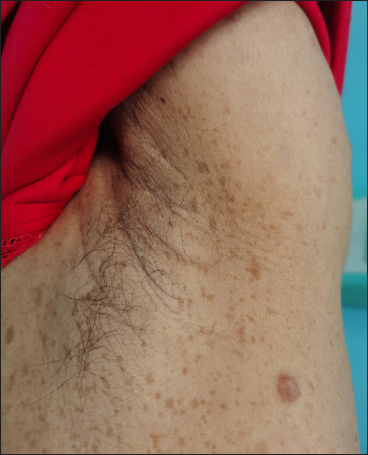
The proband was a 21-year-old woman. There were many café-au-lait macules of different sizes all over the body since one month after birth. The number of the café-au-lait macules in the waist and abdomen gradually increased and fused with age. At around 16-years of age, subcutaneous mass appeared on her both sides of the lumbar ribs and gradually increased in size [Figure 1]. Pathology of subcutaneous mass biopsy showed tumours formed by spindle cells [Figure 1]. neurofibromatosis type 1 was diagnosed by the diagnostic criteria formulated by the National Institutes of Health. The proband’s mother also had café-au-lait macules, tumours and freckles [Figure 2], and a biopsy was performed on her posterior dorsal tumours [Figure 2]. The proband’s maternal grandmother and second aunt also had café-au-lait macules. Her father and sister were normal.
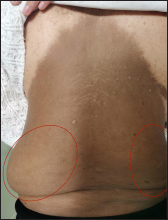
- A 21-year-old woman with neurofibromatosis type 1. (a): Large brown patch on the back of the proband and subcutaneous mass of waist (left about 20 cm * 15 cm * 1 cm, right about 10 cm * 8 cm * 1 cm)
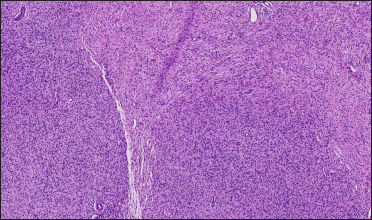
- Histopathology of a subcutaneous mass on the posterior back of the proband (H&E, ×40). Tumours formed by spindle cells were found in the dermis, involving subcutaneous adipose tissue and unclear boundaries. Spindle cell cytoplasm is not obvious, nucleus spindle or oval
- A 52-year-old woman with neurofibromatosis type 1. Cutaneous neurofibroma of various sizes on the back (a) and freckles in the armpit
- A 52-year-old woman with neurofibromatosis type 1. Cutaneous neurofibroma of various sizes on the back (b) of the proband’s mother
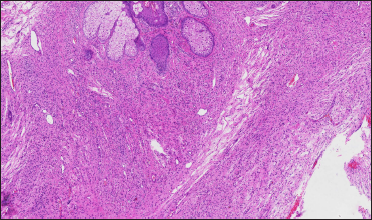
- Histopathology of a cutaneous neurofibroma on the back of the mother (H&E, ×40). The tumours formed by spindle cells have unclear boundaries, unobvious cytoplasm of spindle cells, and spindle or oval nuclei
We collected blood samples from the proband, her parents and sister after the informed consent. This study was approved by the hospital ethics committee. The DNA of the proband’s mother was sent to the company (Shanghai SARO Biotechnology Co., Ltd.) for panel-targeted capture sequencing. The novel mutation was verified in the proband, her father and sister by Sanger sequencing.
A point mutation (c.T1664>G) in exon 15 of the neurofibromatosis type 1 gene (NM_001042492) was found, which caused the cDNA from T to G at position 1664, resulting in premature termination of protein translation at position 555 and abnormal structure of neurofibromin. The mutation was detected in the proband but not in her sister and father by Sanger sequencing [Figure 3]. Mutation Taster predicted this mutation to be “D” (disease causing) and the Likelihood Ratio Test predicts deleterious. It is not described in gnomAD exomes and any other databases. According to the American College of Medical Genetics and Genomics guidelines, this variant is a nonsense mutation and can be interpreted as a pathogenic variant of neurofibromatosis type 1 (PVS1+PM2+PP1).2
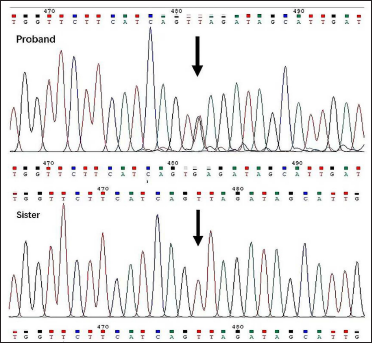
- Nonsense mutation c.T1664>G (position indicated by arrow) in the proband, but not in the sister
More than 3000 pathogenic mutations have been identified in the neurofibromatosis type 1 gene, including missense, nonsense, splicing, microdeletion, microinsertion, total deletion (>20 bp), total insertion (>20 bp) and complex rearrangement. The penetrance of neurofibromatosis type 1 is 100%, but the expression of the neurofibromatosis type 1 gene is highly variable and age-dependent. Even within the same family, the clinical manifestations can be variable in different patients. As in this case, though the proband and her mother held the same mutation, the mother mainly presented with cutaneous neurofibroma, while the proband mainly had giant type plexiform neurofibroma. Phenotype differences may be related to allelic and nonallelic heterogeneity, cell heterogeneity, epigenetics, modified loci, environmental and random factors.
In our case, the mutation is a premature termination codon mutation. Depending on the differences of the clinical manifestations of the proband and her mother, it is not possible to judge whether the mutation causes serious phenotypes, which may require a comprehensive physical examination and long-term follow-up. So far, we have found that five genotypes are clinically identified to be associated with the phenotypes, and there is no correlation between premature termination codon mutations and specific phenotypes. It is only suggested that neurofibromatosis type 1 gene microdeletion and total deletion are associated with severe phenotypes. The number of plexiform and spinal neurofibromas increases, and intellectual and skeletal abnormalities can occur.3 Different phenotypes were found in mutations with different locations. The missense change of codon 1809 (p. Arg1809 Cys) mainly manifested as café-au-lait macules and freckles, macrocephaly, growth retardation and learning problems;4 the missense mutations affecting codons 844–848 are related to the severe phenotypes, which can be manifested as high risk of plexiform neurofibroma, spinal neurofibroma and even malignant transformation.5 Mild phenotype is associated with in-frame deletion of codon 992 (p.Met992del) mainly characterised by pigmentation-like features without cutaneous neurofibroma.6 Therefore, there was no significant phenotypic difference in the mutation types.
In summary, we have identified a novel nonsense mutation in the neurofibromatosis type 1 gene causing different phenotypes of neurofibromatosis in family lines, and our findings expand the spectrum of mutations causing neurofibromatosis type 1 and provide information for genetic counselling.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflict of interest
There are no conflicts of interest.
References
- An epidemiological, clinical and genetic survey of neurofibromatosis type 1 in children under sixteen years of age. Ulster Med J. 2008;77:160-3.
- [PubMed] [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants: Ajoint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype-phenotype correlation in neurofibromatosis type-1: NF1 whole gene deletions lead to high tumor-burden and increased tumor-growth. PLoS Genet. 2021;17:e1009517.
- [CrossRef] [PubMed] [Google Scholar]
- p.Arg1809Cys substitution in neurofibromin is associated with a distinctive NF1 phenotype without neurofibromas. Eur J Hum Genet. 2015;23:1068-71. 10.1038/ejhg.2014.243
- [PubMed] [Google Scholar]
- The natural history of spinal neurofibromatosis: a critical review of clinical and genetic features. Clin Genet. 2015;87:401-10. 10.1111/cge.12498
- [PubMed] [Google Scholar]
- Genotype-phenotype correlations in neurofibromatosis and their potential clinical use. Neurology. 2021;97:S91-8.
- [CrossRef] [PubMed] [Google Scholar]




