
Translate this page into:
A novel mutation in poikiloderma with neutropenia- Clericuzio type in an adult female
Corresponding author: Dr. Vishal Thakur, Department of Dermatology, All India Institute of Medical Sciences, Bathinda, Punjab, India. drvishal87igmc@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Chawla H, Bansal S, Chhabra P, Thakur V. A novel mutation in poikiloderma with neutropenia-Clericuzio type in an adult female. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_1823_2024
Dear Editor,
Poikiloderma with neutropenia-Clericuzio type (PNC) is a rare autosomal recessive disorder characterised by widespread post-inflammatory poikiloderma, pronounced neutropenia and frequent sinopulmonary infections.1 It results from mutations in the USB1 gene, with pathogenic variants like c.531delA, c.496delA, c.179delC and c.326_327del reported to date.2-4 Its exact prevalence remains unknown, with a few cases reported in the literature. Herein, we report a PNC case in an adult female patient with a novel mutation in the USB1 gene.
A 26-year-old woman, born to non-consanguineous parents, presented with hyperpigmented and hypopigmented spots all over the body for seven months of age. She had a history of recurrent sinopulmonary infections associated with fever, malaise and lethargy. However, she denied recurrent cutaneous infections such as recurrent pyoderma, abscess, cellulitis, otitis media, bone involvement or non-healing ulcers. No similar complaints were reported in the family. Physical examination showed a prominent forehead, saddle nose and normal weight and height. Cutaneous examination revealed multiple ill-defined, raindrop-like, hypopigmented, atrophic macules on a background of reticular hyperpigmentation on the trunk [Figure 1a], upper and lower extremities and face [Figure 1b]. There was also a patchy loss of eyebrow hair on the lateral half. Plantar keratoderma [Figure 1c] with normal palms and preserved dermatoglyphics were observed. Toenail dystrophy with mild subungual hyperkeratosis was noticed, while fingernails were normal. Oral examination revealed peg-shaped teeth, loss of multiple molars and premolars, and crowding of central incisors and canines [Figure 1d]. Other mucosa and body hair were normal. Laboratory evaluation showed leukopenia (total leukocyte count: 1500/mm3) with neutropenia (380 cells/mm3). Serum ferritin and lactate dehydrogenase levels were elevated. Bone marrow studies showed no significant abnormality. Other systemic evaluations, including chest X-ray were normal. Whole exome sequencing (WES) identified a homozygous 9-base pair deletion in exon7 of the USB1 gene (c.741_749del variant). Based on the above findings, a diagnosis of PNC was made. This variant was classified as a variant of uncertain significance and had not been previously reported in 1000 Genomes, TopMed, ClinVar, gnomAD, human gene mutation database or online mendelian inheritance in man databases. However, considering the size of the mutation (9 base pair inframe deletion), homozygosity and genotype-phenotype correlation, it was assumed to likely be a novel pathogenic mutation. The patient and her family were counselled for Sanger sequencing and segregation analysis; however, they declined due to financial constraints. She was briefed about the long-term prognosis and advised stringent photo-protection as well as topical keratolytics for plantar hyperkeratosis. Annual complete blood count and dermatological and pulmonary surveillance were advised along with a bi-annual dental examination. Patient was counselled for encouraging at-risk relatives for screening to ensure early diagnosis and prevention of complications.
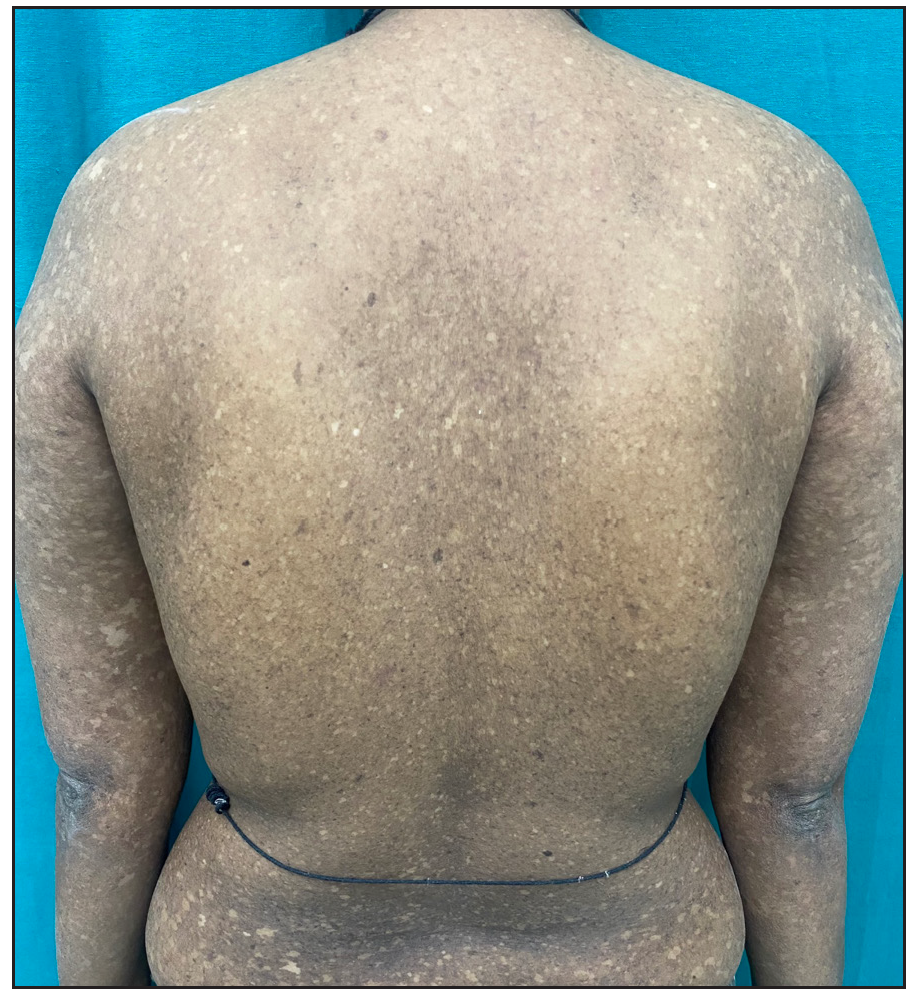
- Multiple ill-defined hypopigmented atrophic macules on a background of reticular hyperpigmentation on the trunk and upper limbs.
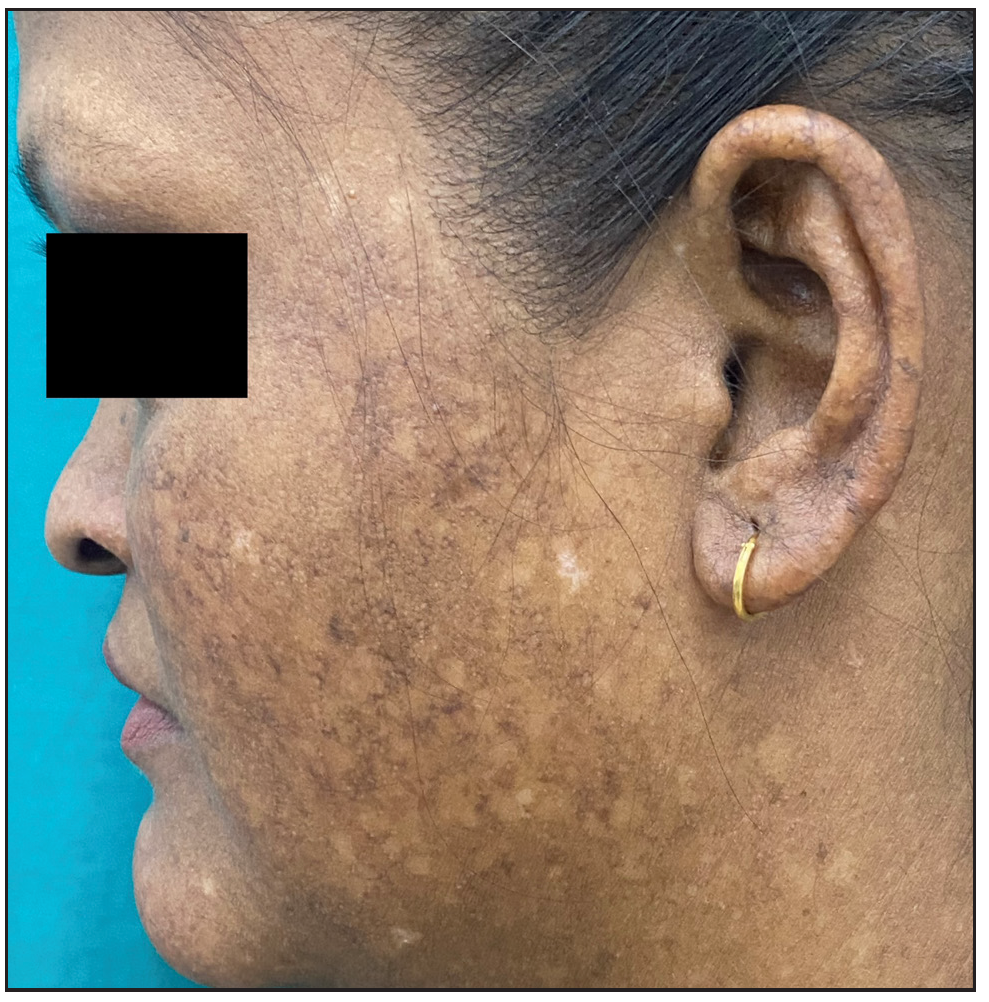
- Multiple ill-defined hypopigmented atrophic macules on a background of reticular hyperpigmentation on left cheek and pinna with partial loss of eyebrow.
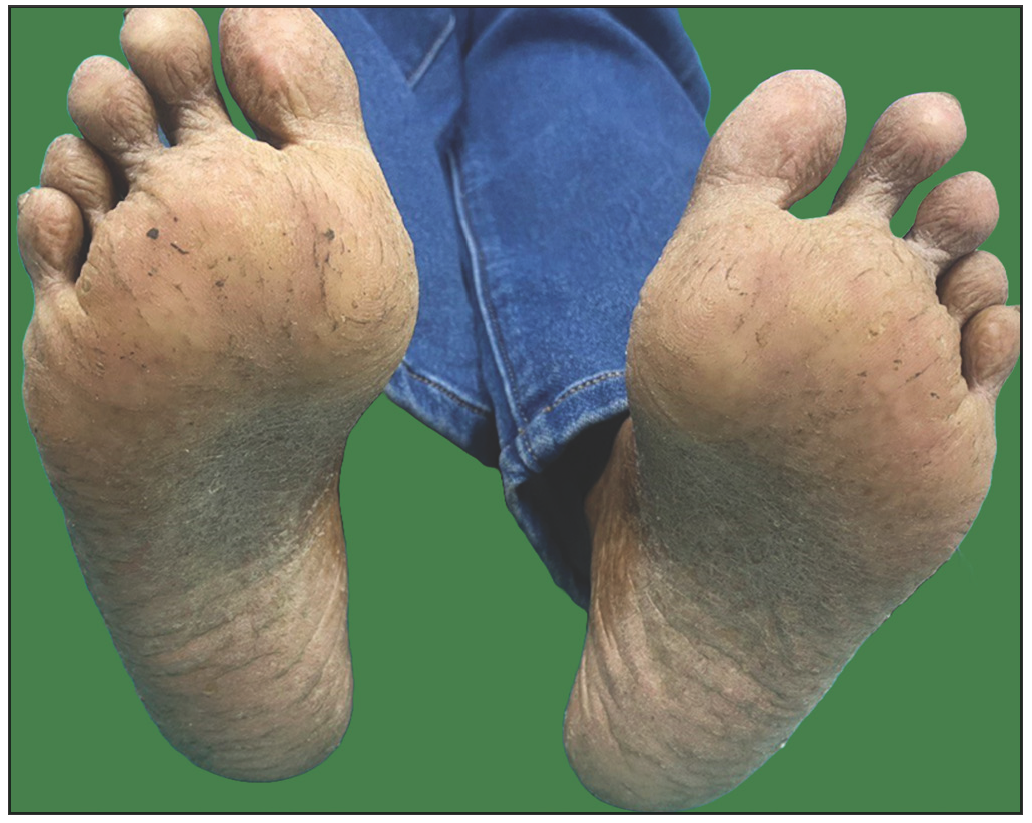
- Clinical image of the patient showing plantar keratoderma.
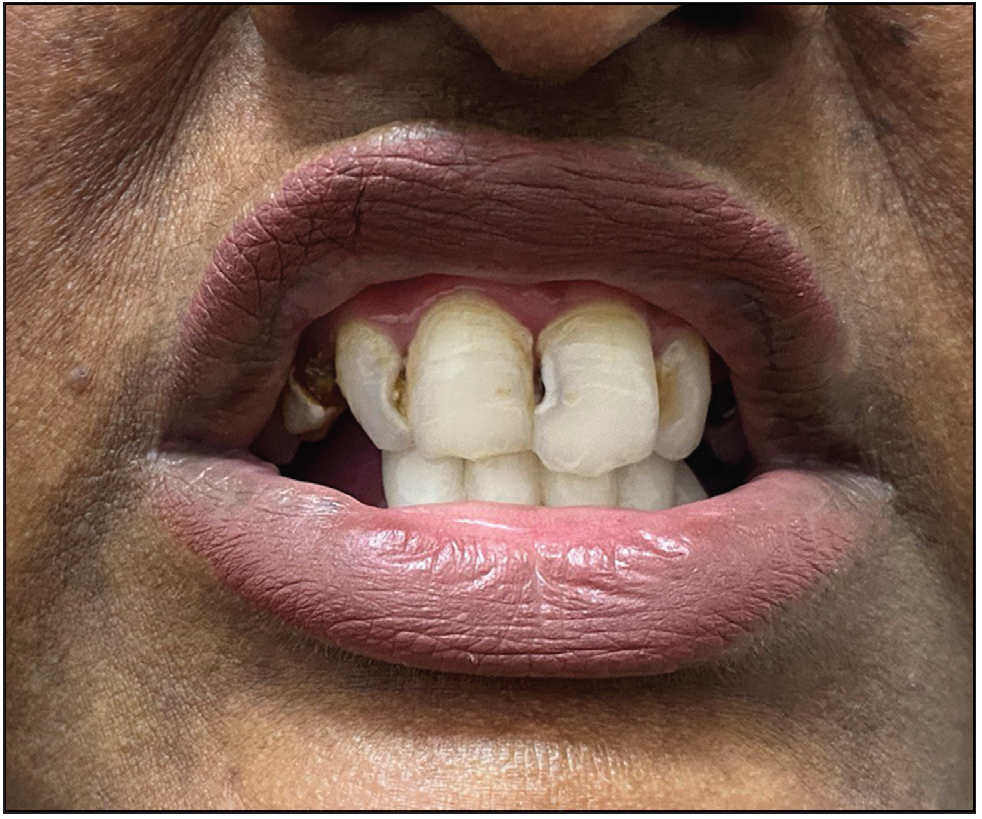
- Clinical image of the oral cavity showing peg-shaped teeth.
PNC must be differentiated from other poikilodermatous disorders, such as Rothmund Thompson syndrome (RTS), Kindler syndrome and dyskeratosis congenita (DKC).5 PNC is characterised by widespread poikiloderma, recurrent infections and cyclic neutropenia due to the mutations in the USB1 gene, which encodes an exonuclease critical for U6 snRNA maturation and spliceosome assembly.3,6 This mutation leads to defective snRNA processing, disrupts haematopoiesis and increases the risk of myelodysplastic syndrome (MDS).3 DKC presents with poikiloderma, oral leukoplakia and dysplastic nails but lacks neutropenia, distinguishing it from PNC. Studies show that PNC can lead to global bone marrow dysfunction affecting all three haematopoietic lineages, increasing the risk of progression to MDS and in some instances, acute myelogenous leukaemia.3 Similarly, individuals with DKC are at an increased risk for bone marrow failure and skin cancers.7 RTS, another close differential, is characterised by poikiloderma, sparse hair, dental and skeletal abnormalities, and an elevated malignancy risk, especially for osteosarcoma and cutaneous malignancies.5 A notable characteristic of RTS is the specific pattern of poikiloderma that begins on the face and extends to the extremities while sparing the trunk. In PNC, it initiates peripherally before progressing to the face and trunk. Another distinguishing trait of RTS is the presence of radial ray abnormalities along with a normal neutrophil count. Managing PNC necessitates a multidisciplinary approach, involving dermatologists, haematologists, geneticists and other specialists for comprehensive care, close surveillance and timely interventions. This case emphasises the importance of revisiting diagnoses in congenital photosensitivity disorders given their considerable phenotypic overlap, thereby highlighting the need of genetic testing for accurate diagnosis. Our report is limited by the absence of in silico analysis and inability to perform Sanger sequencing and segregation analysis in family members for confirming the mutation. However, the identification of a novel variant in the USB1 gene is significant and underscores the ongoing need of genetic research in rare disorders.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Poikiloderma with neutropenia: A novel C16orf57 mutation and clinical diagnostic criteria. Br J Dermatol. 2010;163:866-9.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of a novel C16orf57 mutation in athabaskan patients with poikiloderma with neutropenia. Am J Med Genet A. 2011;155A:337-42.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Insights into mutation effect in three poikiloderma with neutropenia patients by transcript analysis and disease evolution of reported patients with the same pathogenic variants. J Clin Immunol. 2018;38:494-502.
- [CrossRef] [PubMed] [Google Scholar]
- Clericuzio-type poikiloderma with neutropenia in a patient from India. Am J Med Genet A. 2021;185:278-81.
- [CrossRef] [PubMed] [Google Scholar]
- Inherited skin disorders presenting with poikiloderma. Int J Dermatol. 2021;60:1343-5.
- [CrossRef] [PubMed] [Google Scholar]
- USB1 is a miRNA deadenylase that regulates hematopoietic development. Science. 2023;379:901-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The genetics of dyskeratosis congenita. Cancer Genet. 2011;204:635-45.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




