Translate this page into:
A rare case of phakomatosis pigmentovascularis type IIb associated with inverse Klippel–Trenaunay syndrome and Sturge–Weber syndrome
2 Department of Radiology, West China Hospital, Sichuan University, Chengdu, Sichuan, China
Correspondence Address:
Xian Jiang
Department of Dermatovenereology, West China Hospital, Sichuan University, 37 Guoxue Alley, Wuhou District, Chengdu, Sichuan 610041
China
| How to cite this article: Liu L, Sun J, Pan Y, Jiang X. A rare case of phakomatosis pigmentovascularis type IIb associated with inverse Klippel–Trenaunay syndrome and Sturge–Weber syndrome. Indian J Dermatol Venereol Leprol 2019;85:618-620 |
Sir,
A 6-year-old female was brought with port-wine stain, Mongolian spot, nevus anemicus, and atrophy of the right upper and lower limbs which were present since birth. As the child grew, port-wine stain and Mongolian spot faded, but atrophy of the limbs remained. The family history was negative. The port-wine stain was present on the entire face, back of trunk and right extremities, nevus anemicus on the face and Mongolian spot on the trunk [Figure - 1], [Figure - 2], [Figure - 3], [Figure - 4]. The right extremities were about 2.5–3 cm shorter than the left ones and the circumference of the right extremities was about 1.5 cm less than that of the left ones [Figure - 3] and [Figure - 4]. Dermoscopy (DermLite DL3, USA) of the lesions on the face showed red dotted vessels and linear vessels [Figure - 5] representing papillary and subpapillary form of port-wine stain respectively. Magnetic resonance imaging of showed that the diameter of right thigh was about 4.4–8.6 mm less than the left one at corresponding points [Figure - 6]. The cross-sectional area of the subcutaneous adipose tissue of the right thigh was 533 mm [2] less than the corresponding area in the left one, and the area of the muscle in the right thigh was 701.3 mm [2] less than the left [Figure - 7]. Enhanced MRI of head was normal, but electroencephalogram showed frontal spikes that were emitted several times which might be either due to vascular malformations or other causes. Intraocular pressure was 32.1 mmHg in the right eye and 17.7 mmHg in the left. Based on the clinical and radiological manifestations, a diagnosis of phakomatosis pigmentovascularis type IIb associated with inverse Klippel–Trenaunay syndrome and Sturge–Weber syndrome was established. The patient underwent a surgery for glaucoma on the right eye 1 month back following which the intraocular pressure was controlled below 20 mmHg. It was suggested that the patient receive a new therapy known as hematoporphyrin monomethyl ether photodynamic therapy to treat the port-wine stain, but patient's parents were not willing for the same.
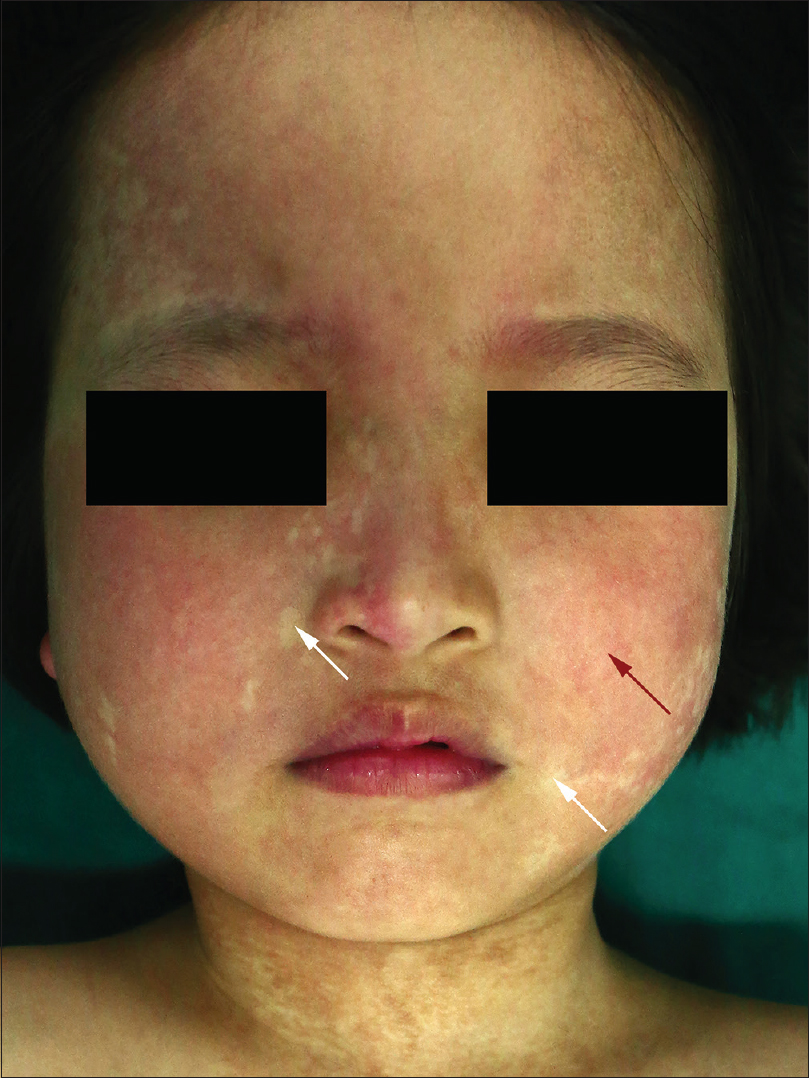 |
| Figure 1: Port-wine stain (red arrow) and nevus anemicus (white arrow) on the face |
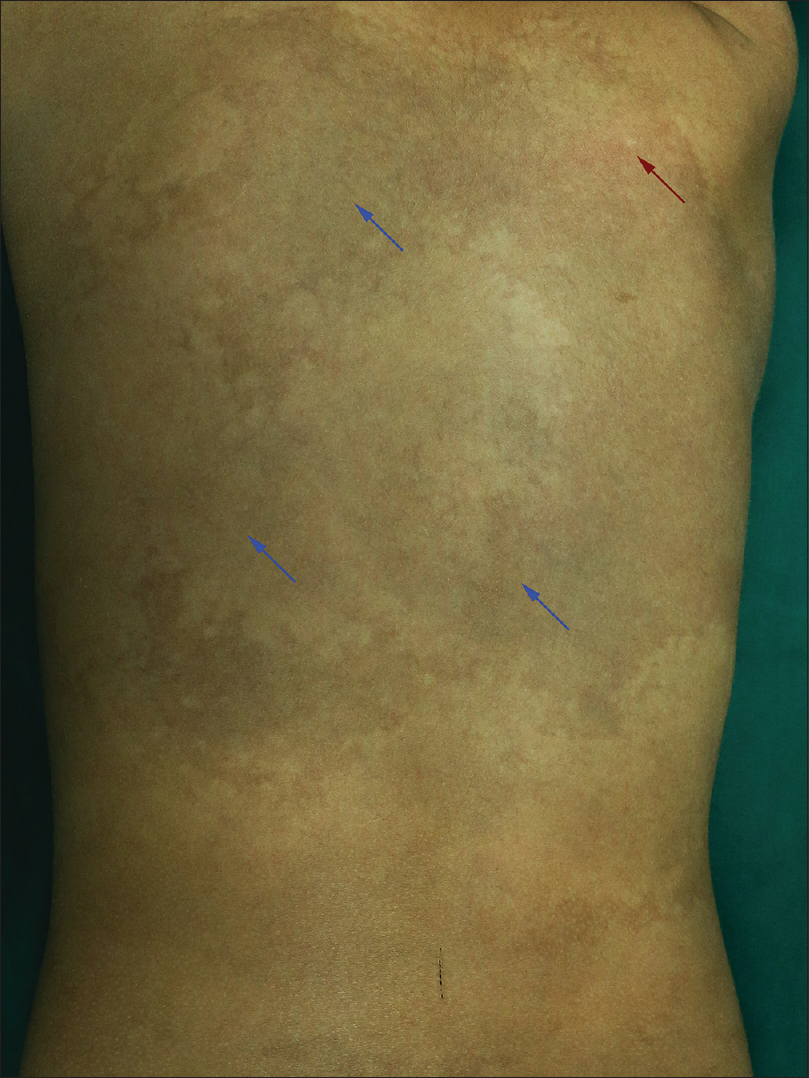 |
| Figure 2: Port-wine stain (red arrow) and Mongolian spot (blue arrow) on the back of trunk |
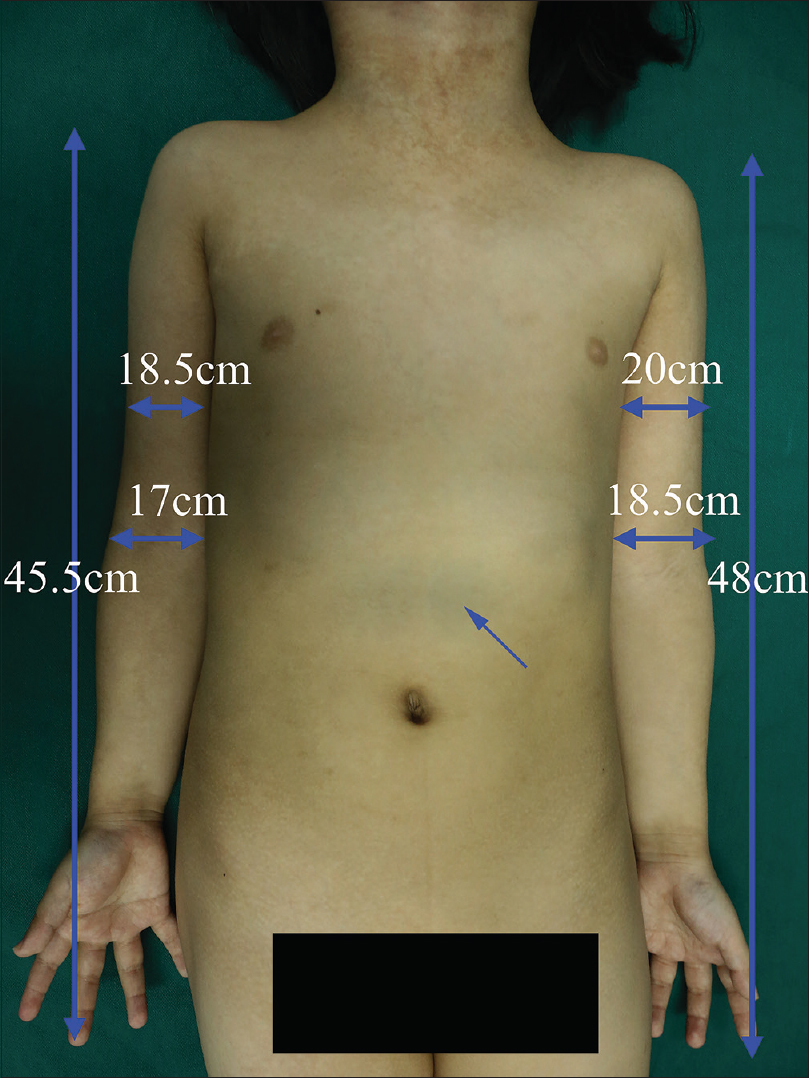 |
| Figure 3: Mongolian spot on chest and stomach; the circumference and length of the right and the left upper extremity |
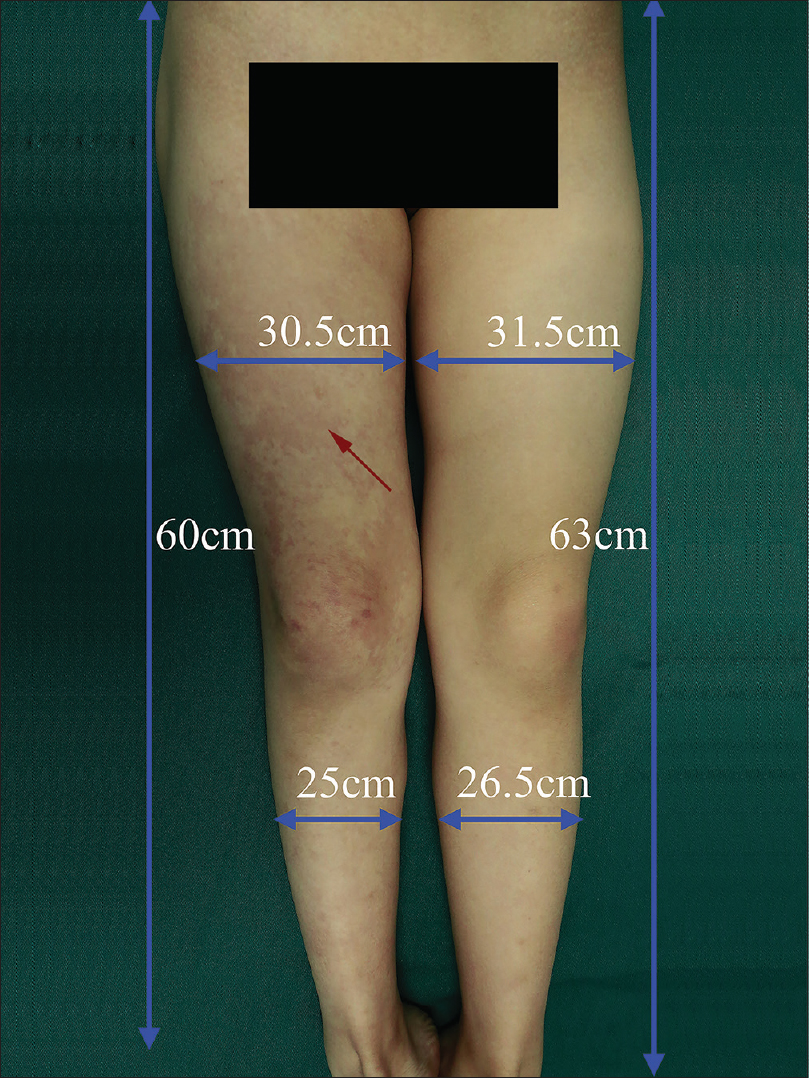 |
| Figure 4: Port-wine stain (red arrow) on the right lower extremity; the circumference and length of the right and left lower extremity |
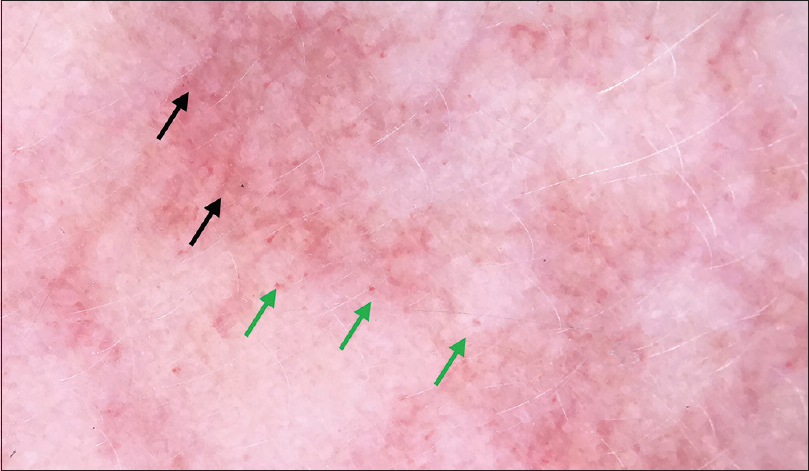 |
| Figure 5: Dermoscopy of port-wine stain of face 200X) showing scattered dotted vessels (green arrow) and linear vessels (black arrow) |
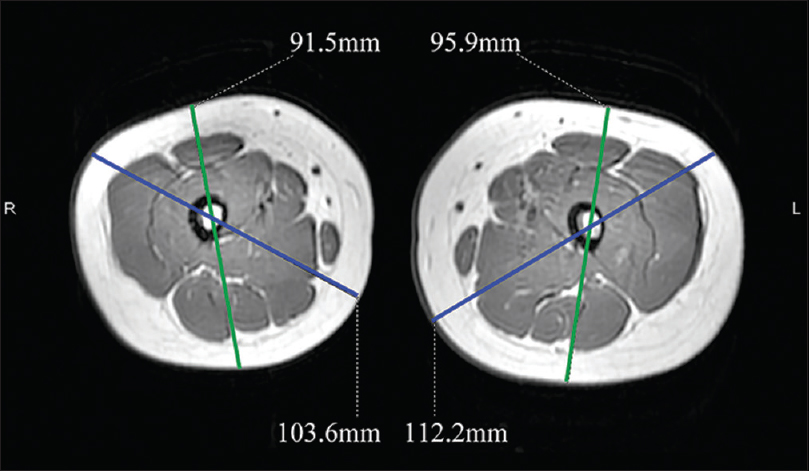 |
| Figure 6: Magnetic resonance imaging; showing the difference in the diameter of right and left thigh |
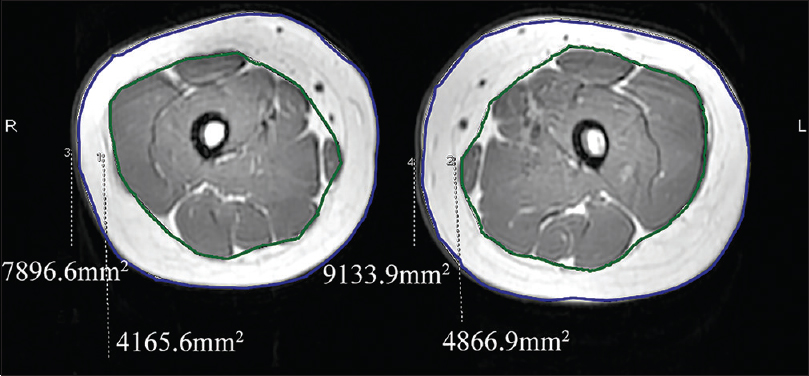 |
| Figure 7: Magnetic resonance imaging showing the difference in the cross-sectional area of subcutaneous adipose tissue and muscle between the thighs |
Even though phakomatosis pigmentovascularis associated with Klippel-Trenaunay syndrome and Sturge-Weber syndrome has been reported, to the best of our knowledge, phakomatosis pigmentovascularis type IIb associated with inverse Klippel-Trenaunay syndrome and Sturge-Weber syndrome has not been reported. Phakomatosis pigmentovascularis is a rare congenital disease involving capillary malformation and pigmented nevi. Type IIb is characterized by capillary malformation and blue spots and systemic signs with or without nevus anemicus. The pathogenesis is still a contentious area. Happle hypothesized that it was a result of genetic mosaicism and allelic mutation called “twin spotting”.[1] Recently, Thomas et al. identified that phakomatosis pigmentovascularis with or without systemic signs was associated with activating a single somatic mutation in GNA11 or GNAQ, encoding Gα subunits of heterotrimeric G proteins. In addition, they found R183C mutation of GNA11 activated p38 MAPK pathway and Q209L mutation of GNA11 activated p38 MAPK, JNK, and ERK pathways in HEK293 cells.[2]
Sturge-Weber syndrome is also a congenital neurocutaneous disease, characterized by capillary malformation and venous capillary abnormalities of the leptomeninges and eyes. The pathogenesis has been reported to be related to the mutation of GNAQ which is the same as phakomatosis pigmentovascularis.[3] ERK activity was increased by R183Q mutation of GNAQ in HEK 293 cells. Klippel-Trenaunay syndrome has a triad of capillary malformations, venous and lymphatic malformations, and bony and/or soft-tissue hypertrophy. If the affected extremities show deficient growth then the condition is known as inverse Klippel-Trenaunay syndrome. Although the mutation of AGGF1 was implicated to be the pathogenesis of Klippel-Trenaunay syndrome, the pathogenesis of inverse Klippel-Trenaunay syndrome is still unclear. The cause of deficient growth is also unknown. It has been speculated that being homozygous for either a 'plus' or a 'minus' allele may be responsible for the over or undergrowth of the affected extremities, respectively.[4] Klippel-Trenaunay syndrome and Sturge-Weber syndrome have already been found together in a patient in whom the tissue from port-wine stain had a somatic mutation of GNAQ.[5] These syndromes may be related to a somatic mutation in genes such as GNAQ or GNA11; however, further studies are required to confirm this hypothesis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the legal guardian has given his consent for images and other clinical information to be reported in the journal. The guardian understands that names and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Happle R. Loss of heterozygosity in human skin. J Am Acad Dermatol 1999;41:143-64.
[Google Scholar]
|
| 2. |
Thomas AC, Zeng Z, Rivière JB, O'Shaughnessy R, Al-Olabi L, St-Onge J, et al. Mosaic activating mutations in GNA11 and GNAQ are associated with phakomatosis pigmentovascularis and extensive dermal melanocytosis. J Invest Dermatol 2016;136:770-8.
[Google Scholar]
|
| 3. |
Shirley MD, Tang H, Gallione CJ, Baugher JD, Frelin LP, Cohen B, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med 2013;368:1971-9.
[Google Scholar]
|
| 4. |
Danarti R, König A, Bittar M, Happle R. Inverse Klippel-Trenaunay syndrome: Review of cases showing deficient growth. Dermatology 2007;214:130-2.
[Google Scholar]
|
| 5. |
Pillai MR, Hasini PP, Ahuja A, Krishnadas SR. A rare case of overlapping Sturge-Weber syndrome and Klippel-Trenaunay syndrome associated with bilateral refractory childhood glaucoma. Indian J Ophthalmol 2017;65:623-5.
[Google Scholar]
|
Fulltext Views
4,527
PDF downloads
1,691





![[Figure - 1]](#fig_ijdvl_2019_85_6_618_268287_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2019_85_6_618_268287_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2019_85_6_618_268287_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2019_85_6_618_268287_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2019_85_6_618_268287_f5.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2019_85_6_618_268287_f6.jpg){kind=link}
![[Figure - 7]](#fig_ijdvl_2019_85_6_618_268287_f7.jpg){kind=link}