Translate this page into:
Advanced type 1 hyperoxaluria presenting as livedo racemosa in a patient with end-stage renal disease
2 Department of Internal Medicine, University and Polytechnic La Fe Hospital, Valencia, Spain
3 Department of Pathology, University and Polytechnic La Fe Hospital, Valencia, Spain
Correspondence Address:
Victor Garcia-Bustos
Department of Internal Medicine, University and Polytecnic La Fe Hospital, Valencia
Spain
| How to cite this article: Torres-Navarro I, Garcia-Bustos V, Botella-Estrada R, Rojas-Ferrer N, Moral-Moral P. Advanced type 1 hyperoxaluria presenting as livedo racemosa in a patient with end-stage renal disease. Indian J Dermatol Venereol Leprol 2019;85:493-495 |
Sir,
Primary hyperoxalurias are rare autosomal recessive disorders which result in precipitation of insoluble oxalate crystals in organs and tissues. Cutaneous calcium oxalate emboli are highly infrequent, and few cases have been published associated with oxalosis. We report the first case of oxalate emboli with a very acute dermatological onset in a patient diagnosed with type 1 hyperoxaluria.
The patient was a 49-year-old Caucasian male with a long history of type 1 primary hyperoxaluria that had been genetically diagnosed at the age of 5 years after recurrent calcium oxalate nephrolithiasis. He was on a combined liver–kidney transplant waiting list because of end-stage renal disease and was undergoing hemodialysis. He presented to our emergency department at the University and Polytechnic La Fe Hospital of Valencia with a 10-hour history of acute leg pain, diffuse reticular rash with palmar-plantar erythema, and violaceous color on the digits [Figure - 1]a and [Figure - 1]b which extended over both the legs [Figure - 1]c and [Figure - 1]d and a 3-week history of distal lower limb paraesthesia. An ambulatory nerve conduction study had previously revealed sensory neuropathy. On examination, the skin showed livedo racemosa as seen in [Figure - 1]a, [Figure - 1]b, [Figure - 1]c, [Figure - 1]d.
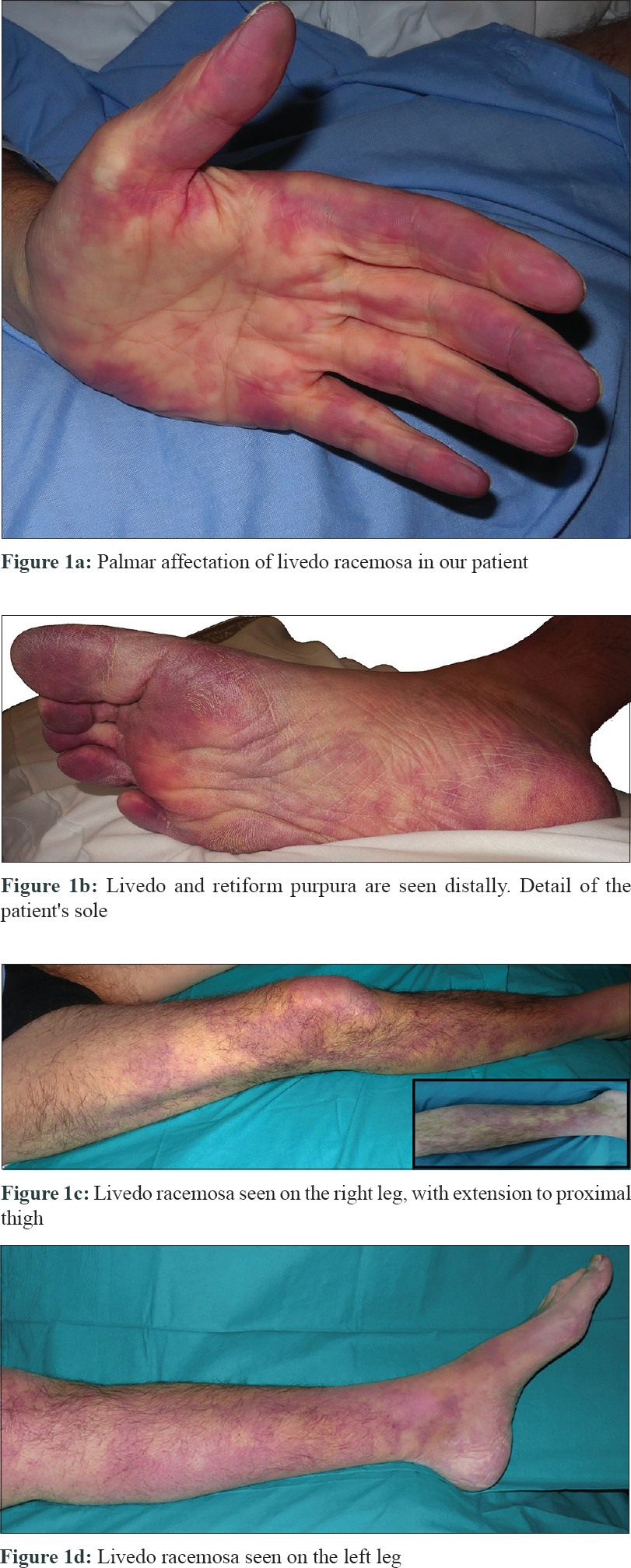 |
| Figure 1 |
Blood test showed leucocytosis [12 × 106/mL, normal range 8–10.5 × 106/mL], highly elevated creatinine (9.28 mg/dL, normal values 0.8–1.1 mg/dL), creatine phosphokinase (2228 U/L, normal 1–7.5 U/L), and C-reactive protein levels (293 mg/L, normal limits 0–5 mg/L). No other alterations were observed.
The differential diagnosis included oxalate embolus, calciphylaxis, cholesterol embolism, uric acid crystal deposition, nephrogenic systemic fibrosis, and other microvascular occlusion syndromes. He had no history of vascular disease, arterial catheterization, or gadolinium exposure and the vascular computerized tomography was normal. Hence, acute arterial occlusion, cholesterol emboli, and nephrogenic systemic fibrosis were ruled out. Thrombophilia study was normal and therefore antiphospholipid syndrome was also discarded. The clinical diffuse appearance did not support the diagnosis of uric acid crystal deposition. Thus, two skin biopsies were taken, one from the palm and the other from the distal region of the leg.
They revealed the presence of abundant birefringent, elongated, diamond-shaped crystals within the dermal and subcutaneous vessels [Figure - 2]a, [Figure - 2]b, [Figure - 2]c, [Figure - 2]d, causing surrounding necrosis. Epidermis was unaltered, and no inflammatory infiltrate was seen. There was no evidence of vascular fibrinoid occlusion, vascular needle-shaped clefts, or calcium deposition. These are the characteristic findings of cutaneous oxalosis and made diagnosis of other microvascular occlusion syndromes unlikely. The patient was admitted to the hospital and intensive daily 6-hour hemodialysis was started. Within the first 24 hours, the reticular erythema improved and the violaceous coloration of both toes disappeared 2 days later. As the symptoms recur early after hemodialysis and oxalate levels may present a rebound elevation of up to 80% of the pre-hemodialysis levels, the patient was discharged under daily intensive hemodialysis.[1] Three months later, his cutaneous lesions persisted. He finally received a combined kidney–liver transplant with complete resolution of the lesions.
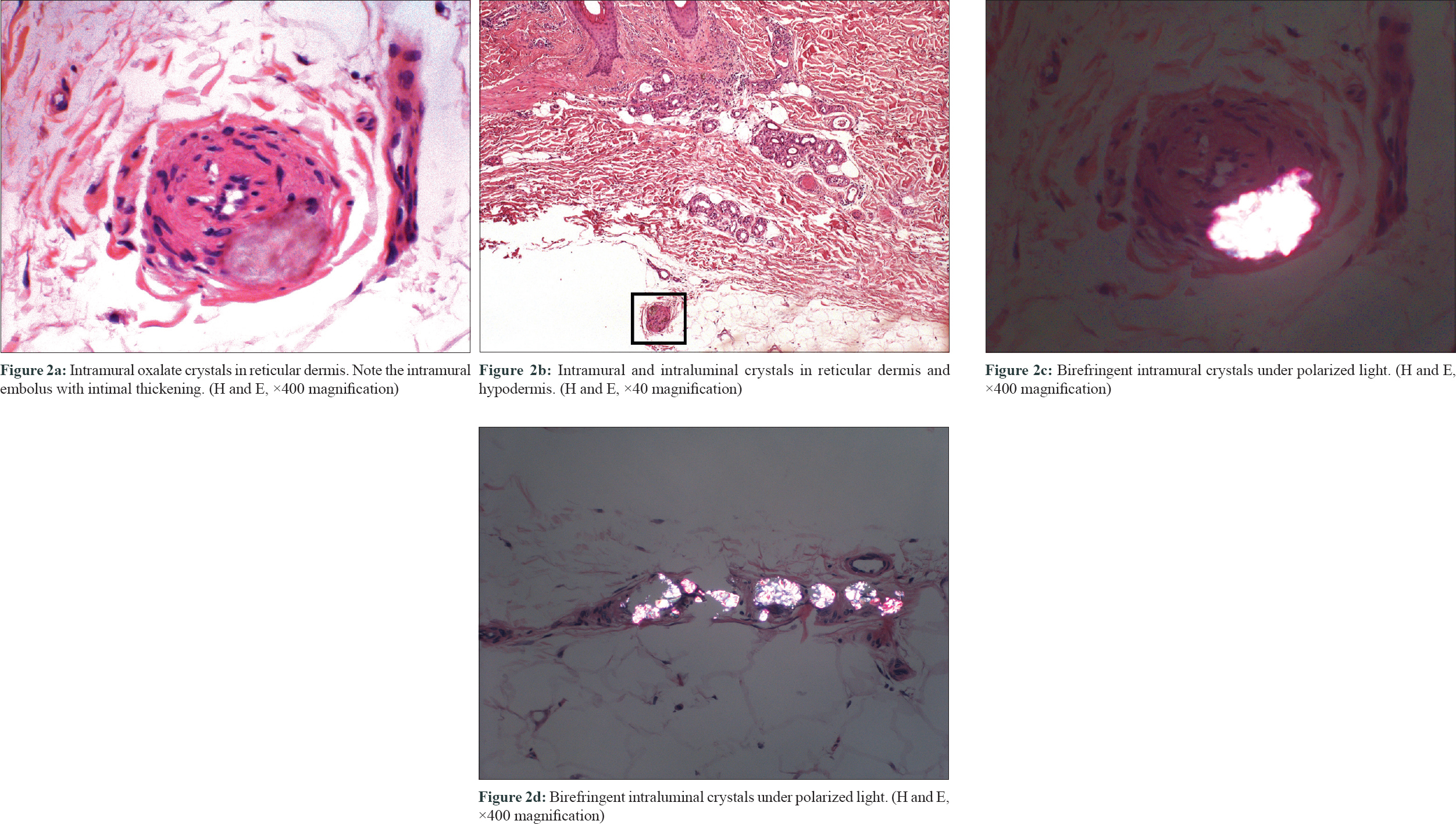 |
| Figure 2 |
Primary hyperoxaluria results from an inborn error of metabolism caused by an enzymatic deficiency that leads to oxalate overproduction and tissue accumulation of oxalate crystals, especially in the kidney.[2] It is divided into three types. Type 1 hyperoxaluria is the most common form and is caused by a defect in a gene on 2q36-q37 encoding alanine-glyoxylate aminotransferase, a hepatic peroxisomal enzyme. It presents with recurrent calcium oxalate nephrolithiasis before the development of renal disease. In primary hyperoxaluria types 2 and 3, end-stage renal disease occurs later and earlier in life, respectively. Secondary hyperoxaluria can occur in patients with increased intestinal absorption or excessive intake of oxalate.[2]
While secondary hyperoxaluria usually results in mild cutaneous disease (acral papules or nodules) because of oxalate extravascular deposition, cutaneous disease associated with primary hyperoxaluria is very rare but severe and diverse.[3],[4],[5] It often manifests with vascular complications such as livedo, acrocyanosis, and peripheral gangrene appearing in the course of several weeks. Most of the lesions are painful.[4] However, no previously reported cases had a sudden onset. It has also been associated with vesicles and blisters,[4] necrotic ulcers due to ischemic necrosis, eschars,[5] and even fibrosis and tenderness [4] which prompts the differential diagnosis with nephrogenic systemic fibrosis. As in our case, the coexistence of peripheral neuropathy and cutaneous oxalosis has also been described.
The most effective treatment is a combined liver–kidney transplant. Liver transplantation is necessary to halt the progression of oxalosis. Both conventional hemodialysis and peritoneal dialysis are not useful to eliminate sufficient quantity of oxalate. However, they have been used to temporarily relieve the symptoms of the cutaneous involvement of hyperoxaluria.[4]
Since patients can present with renal failure and oxalosis before the underlying diagnosis of hyperoxaluria has been made, it is important to consider hyperoxaluria in patients suffering from unexplained soft tissue crystal deposition.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for his images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published, and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Hoppe B, Graf D, Offner G, Latta K, Byrd DJ, Michalk D, et al. Oxalate elimination via hemodialysis or peritoneal dialysis in children with chronic renal failure. Pediatr Nephrol 1996;10:488-92.
[Google Scholar]
|
| 2. |
Cochat P, Rumsby G. Primary hyperoxaluria. N Engl J Med 2013;369:649-58.
[Google Scholar]
|
| 3. |
Winship IM, Saxe NP, Hugel H. Primary oxalosis – An unusual cause of livedo reticularis. Clin Exp Dermatol 1991;16:367-70.
[Google Scholar]
|
| 4. |
Boquist L, Lindqvist B, Ostberg Y, Steen L. Primary oxalosis. Am J Med 1973;54:673-81.
[Google Scholar]
|
| 5. |
Blackmon JA, Jeffy BG, Malone JC, Knable AL Jr. Oxalosis involving the skin: Case report and literature review. Arch Dermatol 2011;147:1302-5.
[Google Scholar]
|
Fulltext Views
2,596
PDF downloads
1,341





![[Figure - 1]](#fig_ijdvl_2019_85_5_493_264159_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2019_85_5_493_264159_f2.jpg){kind=link}