Translate this page into:
Analysis of twenty pediatric cases of tuberous sclerosis complex: Are we doing enough?
2 Department of Skin and VD, Bhimrao Ambedkar Multispecialty Hospital, Sector 30, Noida, Uttar Pradesh, India
Correspondence Address:
Jyoti Nath
House No - 246, Suravi Path, Jatia, Kahilipara Road, Guwahati, Assam - 781 019
India
| How to cite this article: Nath J, Dubey A, Pavan R. Analysis of twenty pediatric cases of tuberous sclerosis complex: Are we doing enough?. Indian J Dermatol Venereol Leprol 2015;81:23-28 |
Abstract
Background: Tuberous sclerosis complex (TSC) is a neuro-cutaneous disease characterized by hamartoma formation in various organs particularly the skin, brain, eye, kidney, heart and lungs. Patients usually have multisystem involvement and thus present to different medical specialties with varied complaints while the true nature of the disease and the hidden manifestations may remain unattended. A im: To assess the frequency and characteristics of various cutaneous and systemic manifestations in TSC and the total impact on different system in a cohort of pediatric patients. Methods: The study included 20 patients fulfilling the diagnostic criteria for TSC from dermatology, pediatrics, and neurology department. Detailed history, examination, and investigations such as chest X-ray, electrocardiography (ECG), ultrasonography (USG) abdomen, echocardiography, fundoscopy, computed tomography (CT) scan of brain and abdomen were done. Results: Dermatological manifestations included ash leaf macules, angiofibromas, shagreen patch, and fibrous plaque. Systemic findings observed were subependymal calcified nodules, subependymal giant cell astrocytoma, cortical tubers, renal cysts, angiomyolipomas, lung cyst, retinal hamartomas, mental deficits and epilepsy. Limitations: Wechsler's Intelligence Scale for Children would have been more appropriate for assessing the intelligence. Dental check-up was not done in our group of patients. Conclusion: Our study highlights the wide variety of cutaneous and systemic manifestations of TSC. The study emphasizes the need for comprehensive multidisciplinary treatment and periodic follow-up which are necessary for appropriate management of this multisystem disorder. Counseling regarding education and rehabilitation of the patients and genetic counseling of parents are important.INTRODUCTION
Tuberous sclerosis complex (TSC) is predominantly a neuro-cutaneous disease characterized by hamartoma formation in multiple organs resulting from defective cellular proliferation and differentiation. [1],[2],[3] It presents with varied cutaneous and systemic features. Cutaneous manifestations include hypomelanotic macules, facial angiofibromas, forehead plaques, shagreen patches, periungual and gingival fibromas, [2] café-au-lait macules, and skin tags. Systemic features include hamartomas in various organs such as cortical tubers, subependymal giant cell astrocytomas, calcified nodules, renal cysts, renal angiomyolipomas, lung cysts, lymphangioleiomyomas, retinal hamartomas, cardiac rhabdomyomas, and nonrenal hamartomas. [2] Initially Virchow and von Recklinghausen identified scleromas and myomata in the brain and heart. [4] Subsequently fibrovascular papules on the face were described by Rayer. [5] In 1908, the classic triad of epilepsy, low intelligence and angiofibromas was proposed by Vogt ("Vogt triad"), which was termed as ′epiloia′ by Sherlock in 1911. [6] In 1920, Van Der Hoeve described retinal hamartomas as ′phakomas′, thus the disease is also known as "phakomatosis". [7] Various studies across the world have reported wide variations in the incidence of the disease. Studies in the pediatric age group reveal an incidence rate of 1 in 5800 at birth and 1 in 15,000 below 5 years of age. [8],[9] Lower incidence rates, up to 1 in 30,000 have been observed in various hospital and population-based studies. [8],[9],[10],[11] It occurs with an equal frequency in males and females.
Though often inherited in an autosomal dominant fashion, approximately 60-70% of cases of tuberous sclerosis are attributed to new mutations. Molecular studies have identified pathogenic mutations in TSC1 or TSC2 genes in 75-90% of patients. [8],[11] However, 10-25% of patients do not show any genetic mutation with currently available testing. In 1993, the TSC2 gene that encodes tuberin and is present on chromosome 16q13, was identified and later TSC1 gene which encodes for hamartin and is present on chromosome 9q34 was identified., [12],[13] Hamartin and tuberin act synergistically to regulate cellular growth and differentiation. [14] Disturbance in this balance leads to interference in organogenesis resulting in hamartoma formation.
Diagnosis is primarily clinical and the first diagnostic criteria were laid down by the first International Tuberous Sclerosis Consensus Conference held in 1998, which was modified in 2004 by Roach and Sparagana. [15],[16] Recently, the second International Consensus Conference in 2012 analyzed the existing criteria with minor changes and stressed the importance of genetic testing, including it in the major criteria. [17] As the manifestations are extremely variable and present in different age groups, the patient visits different specialists as the disease evolves. Thus the total impact of the disease on a patient is not evident till he is thoroughly evaluated and followed up. This study was undertaken to evaluate the characteristics and magnitude of cutaneous and systemic manifestations of TSC patients in the pediatric age group.
METHODS
The study was carried out in Department of Dermatology, Gauhati Medical College, Guwahati, Assam from November 2004 to November 2007. After taking informed consent, patients from the departments of dermatology, pediatrics, neurology, and pediatric surgery fulfilling the revised 2004 criteria for diagnosing tuberous sclerosis complex were included. [16]
Detailed history was taken and mental and psychological status including scholastic performance was grossly assessed while taking history. A thorough clinical examination and relevant investigations were carried out including ECG, echocardiography, ultrasonography, and computed tomography (CT) of abdomen and brain, and X-ray chest, hands, and feet. A thorough ophthalmological examination including fundoscopy was done to look for retinal hamartomas. EEG was performed in relevant cases.
RESULTS
Out of the 20 patients included in the study, 11 were males and 9 were females with a male to female ratio of 1.2:1. The mean age was 11.9 ± 4.8 years (range 5-18 years). Family history was positive in 15% patients, with one set of twin patients.
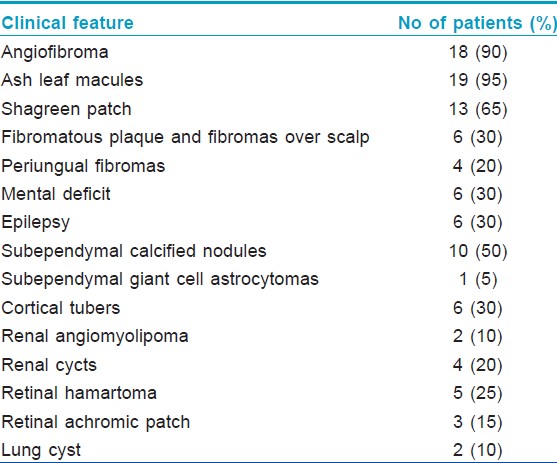
The most common dermatological manifestation in our study was hypopigmented macules observed in 19 (95%) patients followed by angifibromas [Figure - 1] in 18 (90%), shagreen patch in 13 (65%) [Figure - 2], fibromatous plaques and fibromas on scalp in 6 (30%), and periungual fibromas in 4 (20%) patients.
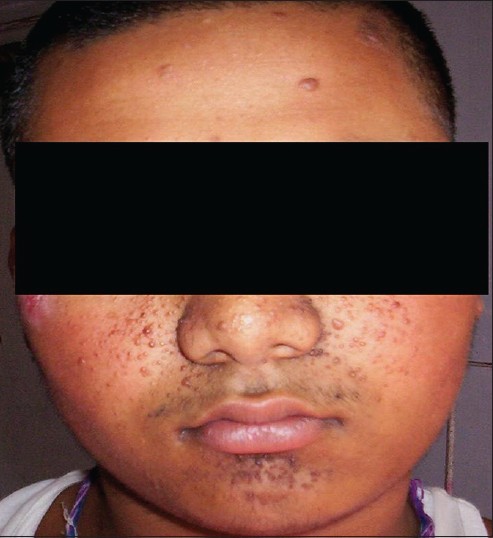 |
| Figure 1: Angiofibromas over the face and forehead plaques |
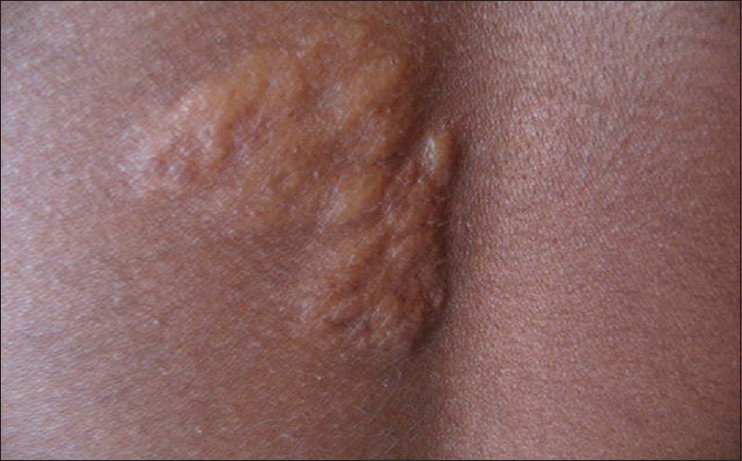 |
| Figure 2: Shagreen patch over the back |
The most common neurological finding in our study was subependymal calcified nodules [Figure - 3] seen in 10 (50%) patients, retinal hamartomas in 8 (40%), mental deficits, epilepsy, cortical tubers, renal cysts and angiomyolipomas [Figure - 4] in 6 (30%) patients each, lung cyst in 2 (10%) and subependymal giant cell astrocytoma [Figure - 5] in 1 (5%) patient.
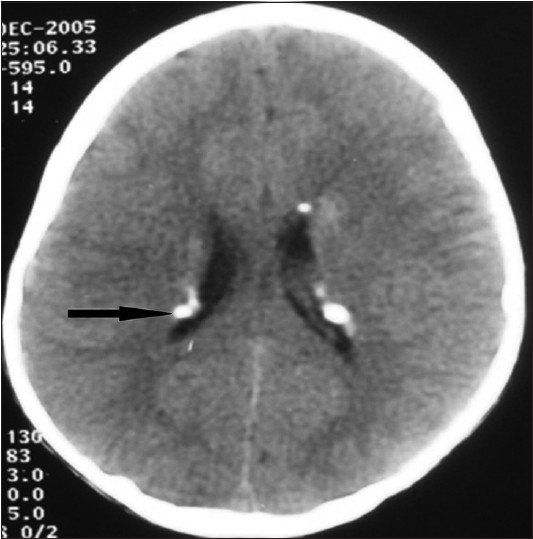 |
| Figure 3: CT brain showing multiple calcified subependymal nodules along the lining of lateral ventricles |
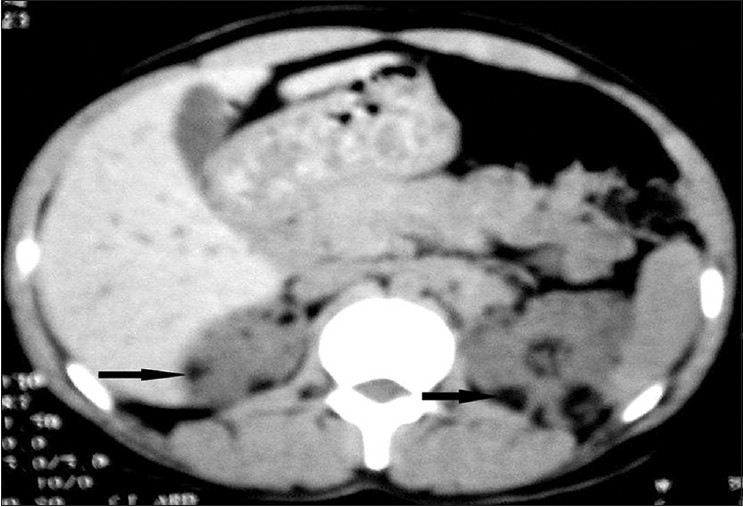 |
| Figure 4: CECT of abdomen showing multiple angiomyolipomas involving bilateral renal cortical parenchyma |
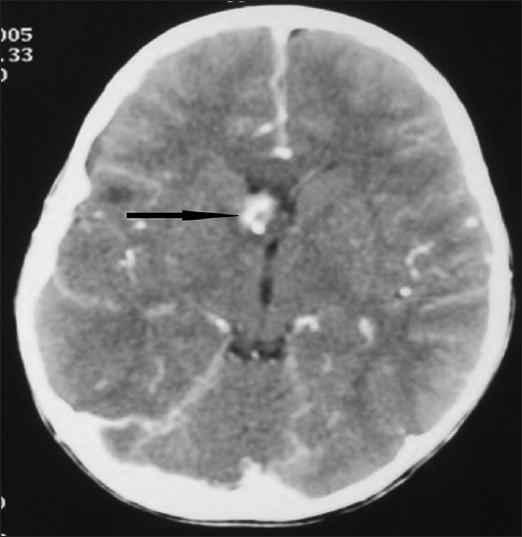 |
| Figure 5: CT brain showing subependymal gaint cell astrocytoma on the right side of foramen of munro |
[Table - 1] shows the cutaneous and systemic manifestations in our patients.
After a thorough analysis, we found all our cases fell into "definite category" of tuberous sclerosis complex with 3-6 major criteria and some minor criteria being fulfilled. Epilepsy and mental retardation are included in the table as they cause considerable impact in the quality of patient′s life even though they are not diagnostic criteria.
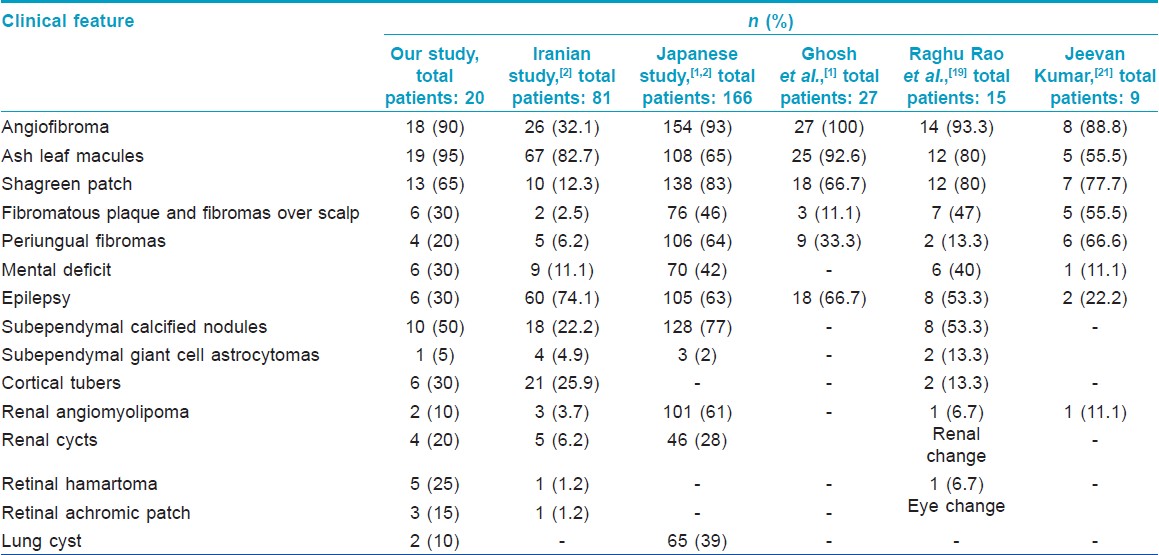
[Table - 2] shows the comparison of various cutaneous and systemic findings of our study with those of other workers.
Limitations
Wechsler′s Intelligence Scale for Children would have been more appropriate for assessing the intelligence. Dental check-up was not done in our group of patients.
DISCUSSION
Before 1998, a diagnosis of tuberous sclerosis was made even when there was a single primary finding such as facial angiofibroma. However, in 1998, diagnostic criteria for the disease were laid down by the Tuberous Sclerosis Alliance at a consensus conference. [4] The diagnostic criteria were further revised in 2004 by Roach and Sparagata. [16] Recently, the second consensus conference in 2012 further simplified and modified the diagnostic criteria and recommended 11 major and 6 minor criteria. The major change was the inclusion of genetic testing, which is taken as a "definite" criterion for the diagnosis irrespective of clinical presentation. [18] All our cases fulfilled 3-6 major criteria and thus fall into the "definite" category. All had hamartomas in multiple organs, which may progress and lead to serious complications in later life.
The earliest cutaneous changes in tuberous sclerosis complex are hypomelanotic macules appearing at birth or during the neonatal period, The macules present in various shapes like ash leaf, confetti-like and polygonal with smooth or irregular margins and the number varies from 1 to 20. Presence of three or more hypomelanotic macules is considered a major criterion and when present at birth should raise a suspicion of tuberous sclerosis complex. Echocardiography should be performed in such patients as the earliest systemic finding is cardiac rhabdomyoma, which develops in utero.
Hypopigmented macules were the most common cutaneous manifestation in our study noted in 95% of the patients. Similarly high incidence of 80%-92.6% was observed in two other Indian studies [1],[18] and a Japanese study, [19] whereas lower incidences of 55.5% and 65% were noted in other studies both in India and abroad. [2],[20]
Facial angiofibromas on centrofacial area are seen in 80% of patients. These erythematous to brown-red papules, nodules, and plaques with a smooth and shiny surface develop by 2 years of age and are fully expressed by adolescence. Previously known as ′adenoma sebaceum′, they are histologically characterized by an irregular proliferation of fibrous tissue and blood vessels and thus the appropriate term is angiofibroma. [21] The incidence of angiofibromas was noted to be high in various studies, ranging from 88% to 100%. [1],[18],[19],[20] We observed angiofibromas in 90% of patients.
Tuberous sclerosis complex also presents with slowly growing, firm, elevated and yellow-brown to skin-colored plaques on the forehead with similar histopathology as facial angiofibromas. [22] The present study shows an incidence of 30% of fibromatous plaques and fibromas over scalp, whereas other studies reported this finding in 2.5-55.5% of patients. [1],[2],[18],[19],[20]
The characteristic skin colored leathery plaques with prominent follicular openings resembling orange peel, usually located in the lumbosacral area are termed shagreen patch. Sixty-five percent of patients had shagreen patch in our study, which was almost similar to the incidence in two other Indian studies (66.7% and 77.7%, respectively). [1],[20] A range of 12.3-83% has been reported in various studies. [1],[2],[18],[19],[20]
Periungual fibromas termed as Koenen tumors start appearing in late childhood as firm, smooth, flesh-colored tumors arising from the nail folds. We found periungual fibromas in 20% of cases, whereas previous reports showed a range of 6.2-66.6%. [1],[2],[18],[19],[20] Other occasional cutaneous findings are café-au-lait macules and skin tags, which were not encountered in our study. [22]
Considering the systemic manifestations of the disease, infantile spasm is the earliest finding. [23] In our series, the youngest patient was 5 years of age and we did not find any case of infantile spasm. Of all the extracutaneous manifestations, subependymal calcified nodule is considered the most common. Multiple calcified subependymal nodules are observed by 2 years of age and are present in almost all patients by puberty. [24] In our study, we found subependymal nodules in 50% of cases while a range of 22.2-53.3% was observed in other studies. [2],[18],[19]
Other neurological features like cortical tubers, subependymal giant cell astrocytomas and cerebral white matter radial migration lines have been described. The latter are caused by failure of a group of neurons to migrate to the area of development. [25] Neural hamartomas clinically manifesting as epilepsy and mental deficiency, learning difficulties, autism, attention deficit disorders and psychosis are often reported. [26],[27],[28] Mental deficit, epilepsy, and cortical tubers were each found in 30% of patients in our series, whereas other studies showed a range of 11.1-42% of mental deficits, [2],[18],[19],[20] 22.2-74.1% of epilepsy, [1],[2],[18],[19],[20] and 13.3-25.9% of cortical tubers [2],[19] Subependymal giant cell astrocytomas were seen in 5% of our patients while a range of 2-13.3% is described in previous reports. [2],[18],[19]
All our patients presenting with epilepsy and mental retardation had one or more neural hamartomas. Basic intelligence was assessed through general interaction and scholastic performance. We feel the incidence might have been recorded to be higher if a standard protocol like Wechsler′s Intelligence Scale for Children had been used. [29]
Tuberous sclerosis complex patients often have retinal lesions of which the most common are retinal hamartomas and retinal achromic patch. Retinal hamartomas have been reported in 1.2-6.7% of patients in other studies, [2],[18] whereas we found a higher incidence of retinal changes in 40% of our cases comprising of retinal hamartomas in 25% and retinal achromic patch in 15%.
Multiple, bilateral renal angiomyolipomas which were asymptomatic in childhood have been observed. Occasionally, renal cell carcinomas have also been encountered. [30] Renal features were found in 30% cases in our series, whereas other studies found a range of 6.2-61%. [2],[18],[19],[20] Of all the renal changes, we found renal cyst in 20% and angiomyolipoma in 10% of cases. We observed lung cysts in 10% of patients, whereas a study from Japan reported an incidence of 39%. [20] Cystic lung disease was seen in 38% of patients with tuberous sclerosis complex [31] and sporadic cases of pulmonary lymphangioleiomyomatosis have been reported. [32],[33]
Cardiovascular manifestations appear early and present as cardiac rhabdomyomas which disappear during adolescence. Arrhythmias and Wolff-Parkinson-White syndrome have been described. [34] We did not find any case of cardiac rhabdomyomas or arrhythmias. Other manifestations like bone cysts have been encountered occasionally. [35]
Though our study revealed a high incidence of extracutaneous manifestations, apart from epilepsy and mental retardation no other complications were observed. However, only long-term follow-up will reveal the progression of the disease process and future complications.
CONCLUSION
It is evident that the incidence rates and manifestations of tuberous sclerosis complex have a wide variation, needing a multicentric study of a large sample size to find out the exact incidence of the disease and its various manifestations. As patients may present to different specialties depending on the symptoms and age, the true nature of the disease may not be apparent and many cases of tuberous sclerosis complex may remain undiagnosed unless the clinician is aware of the disease and its varied manifestations. Hamartomas have a risk of serious consequences if left undiagnosed. There is definite impairment of quality of life of patients and their family members and genetic counseling may prevent the transmission of disease to future offspring. Awareness, interaction, and team work of medical fraternity of different specialties with expertise in the subject may go a long way in recognizing, managing, and preventing future complications in this disease.
| 1. |
Ghosh SK, Debabrata B, Gobinda C, Ghosh A, Sharmila S, Somenath S. Mucocutaneous Changes In Tuberous Sclerosis Complex: A Clinical Profile Of 27 Indian Patients. Indian J Dermatol 2009; 54:255-7.
[Google Scholar]
|
| 2. |
Tonekaboni SH, Tousi P, Ebrahimi A, Ahmadabadi F, Keyhanidoust Z, Zamani GH, et al. Clinical and Para clinical Manifestations of Tuberous Sclerosis: A Cross Sectional Study on 81 Pediatric Patients. Iran J Child Neurol 2012; 6:25-31.
[Google Scholar]
|
| 3. |
Wienecke R, Konig A, Declue JE. Identification of tuberin, the tuberous sclerosis- 2 product. Tuberin possesses specific Rap1 GAP activity. J Biol Chem 1995; 270:16409-14.
[Google Scholar]
|
| 4. |
Pivnik EK, Riccardi VM. The Neurofibromatosis. In: Freedberg IM, Eisen AZ, Wolff K, et al., editors. Fitzpatricks Dermatology in General Medicine. 6 th ed. New York McGraw Hill; 2003. p. 1825-33.
[Google Scholar]
|
| 5. |
Rayer PFO. Treatise: Maladies de la Peau. 2 nd edn. Philadelphia: Williams and Wilkins, 1835. p. 656-9.
[Google Scholar]
|
| 6. |
Sherlock EB. The Feeble-Minded: A guide to st udy and practice. London: MacMillan; 1911. p. 235-47.
[Google Scholar]
|
| 7. |
Gomez MR. Phenotypes of the tuberous sclerosis complex with a revision of diagnostic criteria. Ann NY Acad Sci 1991; 615:1-7.
[Google Scholar]
|
| 8. |
Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci 1991; 615:125-7.
[Google Scholar]
|
| 9. |
Hunt A, Lindenbaum RH. Tuberous sclerosis: A new estimate of prevelance within the Oxford region. J Med Genet 1984; 21:272-7.
[Google Scholar]
|
| 10. |
Wiederholt WC, Gomez MR, Kurland LT. Incidence and prevalence of tuberous sclerosis in Rochester, Minnesota, 1950 through 1982. Neurology 1985; 35:600-3.
[Google Scholar]
|
| 11. |
Sampson JR, Scahill SJ, Stephenson JB, Mann L, Connor JM. Genetic aspects of tuberous sclerosis in the west of Scotland. J Med Genet 1989; 26:28-31.
[Google Scholar]
|
| 12. |
Green AJ, Johnson PH, Yates JR. The tuberous sclerosis gene on chromosome 9q34 acts as a growth suppressor. Hum Mol Genet 1994; 3:1833-4.
[Google Scholar]
|
| 13. |
van Slegtenhorst M, de Hoogt R, Hermans C, Nellist M, Janssen B, Verhoef S, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277:805-8.
[Google Scholar]
|
| 14. |
van Slegtenhorst M, Nellist M, Nagelkarken B, Cheadle J, Snell R, van den Ouweland A, et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 1998; 7:1053-7.
[Google Scholar]
|
| 15. |
Roach ES, Gomez MR, Northrup H. Tuberous Sclerosis Consensus Conference: Revised clinical diagnostic criteria. J Child Neurol 1998; 13:624-8.
[Google Scholar]
|
| 16. |
Roach ES, Sparagana SP. Diagnosis of tuberous sclerosis complex. J Child Neurol 2004; 19:643-9.
[Google Scholar]
|
| 17. |
Northrup H, Krueger DA; International Tuberous Sclerosis Complex Consensus Group. Tuberous Sclerosis Complex Diagnostic Criteria Update: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 2013; 49:243-54.
[Google Scholar]
|
| 18. |
Rama Rao GR, Krishna Rao PV, Gopal K, Kumar YH, Ramachandra BV. Forehead plaque: A cutaneous marker of CNS involvement in tuberous sclerosis. Indian J Dermatol Venereol Leprol 2008; 74:28-31.
[Google Scholar]
|
| 19. |
Wataya-Kaneda M, Tanaka M, Hamasaki T, Katayama I. Trends in the Prevalence of Tuberous Sclerosis Complex Manifestations: An Epidemiological Study of 166 Japanese Patients. PLoS One 2013; 8:e63910.
[Google Scholar]
|
| 20. |
Jeevan kumar B, Thappa DM, Narasimahan R. The cutaneous features of tuberous sclerosis: A hospital based study in south India. Indian J Dermatol 2001; 46:149-53.
[Google Scholar]
|
| 21. |
Nickel WR, Reed WB. Tuberous sclerosis: Special reference to the microscopic alterations in the cutaneous hamartomas. Arch Dermatol 1962; 85:209-26.
[Google Scholar]
|
| 22. |
JóŸwiak S, Schwartz RA, Janniger CK, Micha³owicz R, Chmielik J. Skin lesions in children with tuberous sclerosis complex: Their prevelance, natural course, and diagnostic significance. Int J Dermatol 1998; 37:911-7.
[Google Scholar]
|
| 23. |
Pampiglione G, Moynahan EJ. The tuberous sclerosis syndrome: Clinical and EEG studies in 100 children. J Neurol Neurosurg Psychiatry 1976; 39:663-73.
[Google Scholar]
|
| 24. |
Jozwiak S, Schwartz RA, Janniger CK, Bielicka-Cymerman J. Usefulness of diagnostic criteria of tuberous sclerosis complex in paediatric patients. J Child Neurol 2000; 15:652-9.
[Google Scholar]
|
| 25. |
Curatolo P, Cusmai R, Cortesi F, Chiron C, Jambaque I, Dulac O. Neuropsychiatric aspects of tuberous sclerosis. Ann NY Acad Sci 1991; 615:8-16.
[Google Scholar]
|
| 26. |
Hunt A, Dennis J. Psychiatric disorder among children with tuberous sclerosis. Dev Med Child Neurol 1987; 29:190-8.
[Google Scholar]
|
| 27. |
Smalley SL, Tanguay PE, Smith M, Gutierrez G. Autism and tuberous sclerosis. J Autism Dev Disord 1992; 22:339-55.
[Google Scholar]
|
| 28. |
Roach ES, DiMario FJ, Kandt RS, Northrup H. Tuberous sclerosis consensus conference: Recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol 1999; 14:401-7.
[Google Scholar]
|
| 29. |
Wechsler D. Wechsler Intelligence Scale for Children-4 th Edition (WISC-IV®). San Antonio, TX: Harcourt Assessment; 2003.
[Google Scholar]
|
| 30. |
Behnes CL, Schütze G, Engelke C, Bremmer F, Gunawan B, Radzun HJ, et al. 13-year-old tuberous sclerosis patient with renal cell carcinoma associated with multiple renal angiomyolipomas developing multifocal micronodular pneumocyte hyperplasia. BMC Clin Pathol 2013; 13:4.
[Google Scholar]
|
| 31. |
Ryu JH, Sykes AM, Lee AS, Burger CD. Cystic lung disease is not uncommon in men with tuberous sclerosis complex. Respir Med 2012; 106:1586-90.
[Google Scholar]
|
| 32. |
Silva OR, Puelma CF, Retamal PV, Rojas Sm P, Cruzat CC, Reyes GC. Pulmonary lymphangioleiomyomatosis: Report of one case. Rev Med Chil 2009; 137:1474-7.
[Google Scholar]
|
| 33. |
Lu SH, Hou YY, Tan YS, Xu JF, Zeng HY, Sujie AK, et al. Clinical and histopathological alterations of lymphangioleiomyomatosis in 14 Chinese patients. Chin Med J (Engl) 2009; 122:1895-900.
[Google Scholar]
|
| 34. |
O'Callaghan FJ, Clarke AC, Joffe H, Keeton B, Martin R, Salmon A, et al. Tuberous sclerosis complex and Wolff-Parkinson-White syndrome. Arch Dis Child 1998; 78:159-62.
et al. Tuberous sclerosis complex and Wolff-Parkinson-White syndrome. Arch Dis Child 1998; 78:159-62.'>[Google Scholar]
|
| 35. |
Fraioli MF, Lecce M, Fraioli C, Paolo C. Spinal gaint cell tumor in tuberous sclerosis: Case report and review of the literature. J Spinal Cord Med 2013; 36:157-60.
[Google Scholar]
|
Fulltext Views
7,069
PDF downloads
3,678





![[Figure - 1]](#fig_ijdvl_2015_81_1_23_148560_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_1_23_148560_f3.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_1_23_148560_f4.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2015_81_1_23_148560_f5.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2015_81_1_23_148560_f6.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2015_81_1_23_148560_t2.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2015_81_1_23_148560_t7.jpg){kind=link}