Translate this page into:
Anti-RO 52-positive systemic sclerosis sine scleroderma with multisystem involvement and recurrent vasculitis
Correspondence Address:
Angoori Gnaneshwar Rao
F12, B8 Hig.2 APHB, Baghlingampally, Hyderabad - 500 044, Telangana
India
| How to cite this article: Rao AG, Farheen SS, Reddy UD, Kolli A, Aparna K, Kranthi J, Haqqani R, Rani T S. Anti-RO 52-positive systemic sclerosis sine scleroderma with multisystem involvement and recurrent vasculitis. Indian J Dermatol Venereol Leprol 2018;84:607-610 |
Sir,
Systemic sclerosis sine scleroderma was first described by Rodnan and Fennell in 1962.[1] It is a rare subgroup of scleroderma accounting for 2%–9% of scleroderma patients and is characterized by internal organ involvement and absence of scleroderma, noted in other forms of systemic sclerosis.[2]
A 25-year-old lady presented to our hospital with recurrent asymptomatic eruption on upper limbs, chest and legs of 5 months duration. There was no history of fever, abdominal pain, joint pain and photosensitivity. She gave a history of developing pericardial effusion for which she underwent pericardiocentesis 1 year back. Cutaneous examination revealed pinchable skin on metacarpophalangeal joints. Multiple discrete depigmented atrophic shiny macules were distributed on both upper limbs, chest, upper back, knees and dorsa of feet. A few skin colored papules with central crusting were also noted among atrophic macules [Figure - 1],[Figure - 2],[Figure - 3]. The right index finger and middle finger showed dry gangrene with bluish discoloration [Figure - 4]. A single, large, round punched-out ulcer of 2 cm in diameter was present on the dorsum of the right foot, covered with necrotic slough [Figure - 5]. Radial arterial pulsations on both sides were absent. A provisional diagnosis of vasculitis with peripheral vascular disease was made. Dego’s disease was considered as a differential diagnosis. Routine hematological and biochemical investigations were unremarkable. Antinuclear antibody, anti-ds DNA, anti-Scl 70 antibodies, anticentromere antibodies, anti-polymerase III antibodies, U1 RNP antibodies, anticardiolipin antibodies, cytoplasmic antineutrophil cytoplasmic antibody and perinuclear anti-neutrophil cytoplasmic antibody were negative. Anti-RO52 antibodies were positive. Rheumatoid factor, venereal research laboratory test and serology for human immune deficiency virus were non-reactive. Chest skiagram showed pulmonary hypertension. Barium swallow and ultrasonography of abdomen revealed normal study. Color Doppler study of both upper limbs revealed low systolic and diastolic flow velocities with a monophasic spectral pattern. Computerized tomographic scan of chest showed ground glass opacities predominantly involving lower lobes and a few pulmonary nodules in right upper and lower lobes suggestive of interstitial lung disease and pulmonary arterial hypertension. Two-dimensional echocardiography also showed pulmonary arterial hypertension. Histopathological examination of biopsy from a representative papule showed perivascular collections of neutrophils, eosinophils and lymphocytes in dermis. The vessel wall showed transmural inflammation, suggestive of vasculopathy [Figure - 6]. Biopsy from the ulcer showed inflammatory neutrophilic infiltrate, fibrinoid necrosis and endothelial proliferation, suggestive of vasculitis [Figure - 7]. Alcian blue stain for mucin was negative. The absence of characteristic clinical features of scleroderma, absence of anti-Scl 70 and anti-centromere antibodies, and positive imagological evidence of pulmonary arterial hypertension, interstitial lung disease and peripheral vascular disease, along with features of vasculitis on histopathology of skin biopsy corroborated in establishing the diagnosis of systemic sclerosis sine scleroderma with interstitial lung disease, pulmonary arterial hypertension, peripheral vascular disease and vasculitis. Dego’s disease was considered as a differential diagnosis as the skin lesions showed atrophy with shiny surface involving typical sites and associated multisystem involvement.
 |
| Figure 1: Atrophic, depigmented macules on right arm |
 |
| Figure 2: Atrophic, depigmented macules on left elbow |
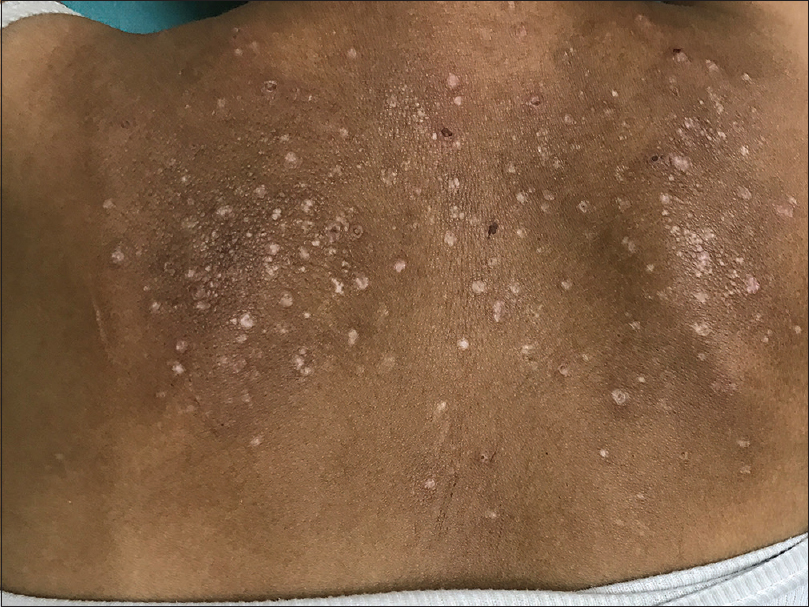 |
| Figure 3: Atrophic depigmented macules on the back |
 |
| Figure 4: Dry gangrene of right index finger and middle finger with bluish discoloration |
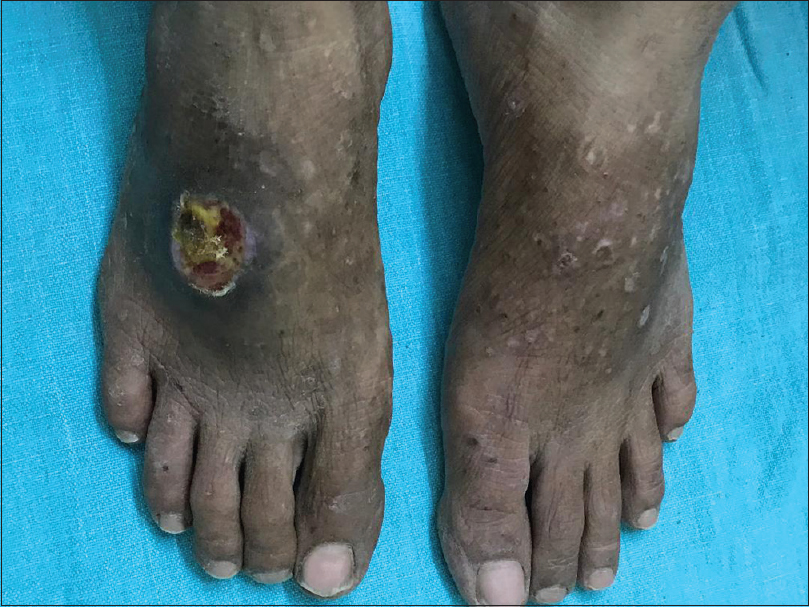 |
| Figure 5: Punched out ulcer on the dorsum of right foot with vasculitic lesions and atrophic depigmented macules |
 |
| Figure 6: Perivascular collections of neutrophils, eosinophils and lymphocytes in dermis with transmural inflammation of the vessel wall (H and E, ×400) |
 |
| Figure 7: Inflammatory neutrophilic infiltrate, fibrinoid necrosis and endothelial proliferation in the small vessel wall (H and E, ×400) |
Systemic sclerosis sine scleroderma is a rare variant of scleroderma characterized by internal organ involvement and absence of scleroderma. The episode of pericardial effusion 1 year back could be an early sign of systemic sclerosis sine scleroderma in the index case. Subsequent development of pulmonary arterial hypertension, interstitial lung disease and peripheral vascular disease validates the diagnosis of systemic sclerosis sine scleroderma which is further substantiated by the absence of sclerodermatous changes in the histopathology of skin and absence of specific antibodies such as anti-Scl 70 and anti-centromere antibodies. The atrophic cutaneous lesions with shiny surface prompted us to consider the diagnosis of Dego’s disease, but the absence of clinical and imagological evidence of involvement of gastrointestinal and central nervous system could not support the diagnosis. Moreover, the absence of wedge-shaped connective tissue necrosis and lack of mucin deposition on histopathological examination characteristic of Dego’s disease could not substantiate the diagnosis.[3] It is known that 50% of these patients have involvement of gastrointestinal system with intestinal perforation as the most severe complication. Central nervous system is involved in 20% of them and manifestations usually develop weeks to years after the onset of cutaneous lesions.
The absence of characteristic sclerodermatous features in our case is an important factor for the diagnosis of systemic sclerosis sine scleroderma. Interestingly, in a study by Poormoghim et al., 18 cases grouped as systemic sclerosis sine scleroderma, subsequently, developed sclerodactyly during the follow-up period of 3.9 years. Moreover, they consider systemic sclerosis sine scleroderma to be part of the continuance of limited systemic sclerosis rather than a separate entity.[2] Similarly, a long-term follow-up may determine whether the index case is systemic sclerosis sine scleroderma or a part of the continuance of limited cutaneous form of systemic sclerosis. The predominant systems involved in systemic sclerosis sine scleroderma are pulmonary, gastrointestinal and peripheral vascular system. Notably, there is pulmonary involvement in the index case. Similarly, a Brazilian study on systemic sclerosis sine scleroderma reported pulmonary involvement in 63.2% of cases. Moreover, the same study revealed esophageal involvement in 83.1% of cases. However, the esophagus was not involved in the index case. Pulmonary arterial hypertension is a major cause of mortality in systemic sclerosis sine scleroderma and can be a presenting feature of systemic sclerosis sine scleroderma.[4] Incidentally, pulmonary arterial hypertension was detected in the index case. Digital ischemia has been reported in association with anticardiolipin antibodies; however, the digital gangrene involving index finger and middle finger in the absence of anticardiolipin antibodies in the index case is noteworthy.[5]
The occurrence of recurrent cutaneous vasculitis in the index case is inexplicable and we were unable to find previous report of association of systemic sclerosis sine scleroderma with cutaneous vasculitis. Nonetheless, antineutrophil cytoplasmic antibody–associated vasculitis has been reported in association with systemic sclerosis combined with glomerulonephritis.[6]
Interestingly, the index case was positive for anti-RO52 antibodies. Menéndez et al., while analyzing the diagnostic utility of anti-RO52 antibodies in autoimmune diseases, found it positive in all cases (100%) of dermatomyositis and polymyositis and 80% of diffuse systemic sclerosis cases. They further opined that isolated detection of anti-RO52 antibodies probably precede the development of autoimmune disease. Incidentally, they observed that the pulmonary manifestations were often associated with anti-RO52 antibodies.[7] Likewise, interstitial lung disease in the index case may be attributed to the presence of anti-RO52 antibodies.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Rodnan GP, Fennell RH Jr. Progressive systemic sclerosis sine scleroderma. JAMA 1962;180:665-70.
[Google Scholar]
|
| 2. |
Poormoghim H, Lucas M, Fertig N, Medsger TA Jr. Systemic sclerosis sine scleroderma: Demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum 2000;43:444-51.
[Google Scholar]
|
| 3. |
Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Köhlmeier-Degos disease) – A review. Orphanet J Rare Dis 2013;8:10.
[Google Scholar]
|
| 4. |
Pauling JD, Gunawardena H, Coghlan JG, Easaw J, Suntharalingam J, McHugh NJ, et al. Pulmonary artery hypertension as the presenting feature of systemic sclerosis sine scleroderma. Rheumatology (Oxford) 2008;47:1431-2.
[Google Scholar]
|
| 5. |
Herrick AL, Oogarah PK, Freemont AJ, Marcuson R, Haeney M, Jayson MI, et al. Vasculitis in patients with systemic sclerosis and severe digital ischaemia requiring amputation. Ann Rheum Dis 1994;53:323-6.
[Google Scholar]
|
| 6. |
Omair MA, Mohamed N, Johnson SR, Ahmad Z, Lee P. ANCA-associated vasculitis in systemic sclerosis report of 3 cases. Rheumatol Int 2013;33:139-43.
[Google Scholar]
|
| 7. |
Menéndez A, Gómez J, Escanlar E, Caminal-Montero L, Mozo L. Clinical associations of anti-SSA/Ro60 and anti-ro52/TRIM21 antibodies: Diagnostic utility of their separate detection. Autoimmunity 2013;46:32-9.
[Google Scholar]
|
Fulltext Views
4,812
PDF downloads
1,445





![[Figure - 1]](#fig_ijdvl_2018_84_5_607_238093_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2018_84_5_607_238093_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2018_84_5_607_238093_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2018_84_5_607_238093_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2018_84_5_607_238093_f5.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2018_84_5_607_238093_f6.jpg){kind=link}
![[Figure - 7]](#fig_ijdvl_2018_84_5_607_238093_f7.jpg){kind=link}