Translate this page into:
Apert syndrome
Correspondence Address:
Mahru Tajalli
Department of Dermatology, Emam Khomeini Hospital, 61335, Ahwaz
Iran
| How to cite this article: Yaghoobi R, Bagherani N, Tajalli M, Paziar N. Apert syndrome. Indian J Dermatol Venereol Leprol 2010;76:724 |
Sir,
Apert syndrome (AS) (acrocephalosyndactyly type I) is a rare malformation syndrome first described by Wheaton in 1894, [1] and later by the French physician Eugλne Apert. [1],[2],[3]
Incidence of AS is approximately one in 50,000 birth. [2],[3],[4] Some investigators state that 4.5% of all craniosynostosis represent AS. [2],[4]
A 23-year-old man presented with the complaint of severe acne vulgaris, involving the face, trunk and extremities, for six years. He was also noted to have developmental abnormalities including skeletal malformations as short stature (the height of 153 cm), lordosis, kyphosis and bilateral scapula hypoplasia especially in the right side, symmetric shortening of all digits of the hands and feet, disfigurement of the face and head as macrocephaly, acrocephaly, flattened occiput, ocular hypertelorism, proptosis, mid face hypoplasia, incomplete dentition, lumbosacral hypertrichosis (faun tail), unilateral lymphedema in the left side, and gynecomastia. Also, he had light-colored skin and eyes. These manifestations helped in the diagnosis of Apert syndrome [Figure - 1].
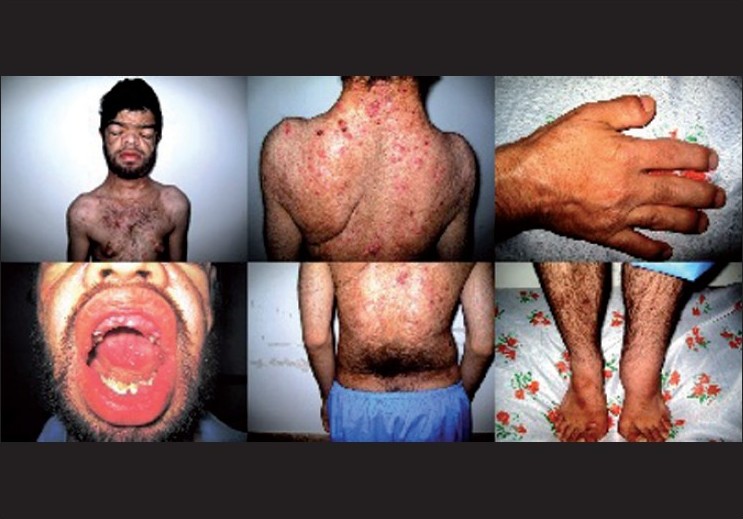 |
| Figure 1 :A 23-year-old man, diagnosed with Apert syndrome, with acrocephaly, mid face hypoplasia, gynecomastia and many acne vulgaris- attributed lesions, scapula hypoplasia, symmetric brackydactyly of all digits of the hands, incomplete dentition, lumbosacral hypertrichosis appearing as faun tail, and left-sided lymphedema |
He had a history of growth and developmental retardation in infancy, but intelligence level was normal. The patient had undergone cardiac surgery for repairing patent ductus arteriosus (PDA) in childhood.
He was the first child in his family with six members. His eight-month-old sister was also afflicted with such manifestations [Figure - 2]. Her intelligence level was also normal. His parents were consanguineous (second-degree), and had no history of any developmental and genetic disorders.
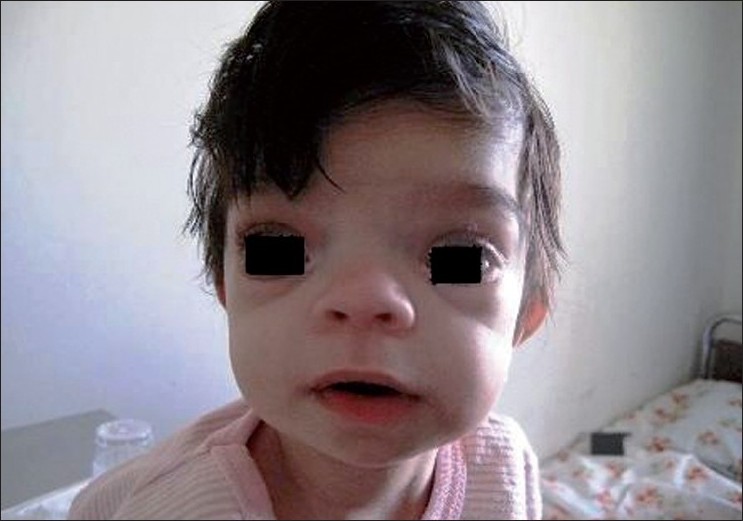 |
| Figure 2 :An eight-month-old baby, sister of the patient shown in Figure 1, with mid face hypoplasia and shallow orbit compatible with the diagnosis of Apert syndrome |
Abdominal ultrasound revealed absence of splenomegaly. In echocardiography, a decreased ejection fraction was detected. Brain computed tomography, which had been performed many years ago, showed no abnormality.
AS is characterized by craniosynostosis associated with maxillary hypoplasia, symmetric syndactyly of the hands and feet (which minimally involves digits 2, 3, and 4), and other systemic malformations including mental retardation. [1],[4]
Mutation in the fibroblast growth factor receptor 2 gene (FGFR2) maps to chromosome 10q25-10q26 cause AS. [5],[7] It is known to be inherited in an autosomal dominant fashion [1],[2],[4],[6],[7],[8] as well as germinal mosaicism, [1] but more cases are sporadic. [1],[2],[4],[5],[6],[7] The sporadic cases are postulated to be associated with advanced paternal age. [2],[4],[7] Recurrence risk for an affected individual to have an affected offspring is 50%. [5] In view of inheritance pattern in AS and considering the involvement of two members in our patient′s family and the parents being healthy, it appears that mosaicism in paternal gonads is the best explanation for the inheritance pattern in our patient.
AS is thought to occur as a result of androgen end-organ hyper-response affecting the epiphyses and sebaceous glands. [5],[7] This results in early epiphyseal fusion, resulting in short stature, short and fused digits, and acrocephaly. [5]
The common skin manifestations of AS include hyperhidrosis, brittle nails, and synonychia. This combination often leads to candidal infection and colonization. [4] The dermatologic hallmark of AS is severe inflammatory and comedonal acne involving the face, chest, back, and also unusual areas such as the forearms, buttocks, and thighs. [6] The rate of sebum secretion correlates with acne severity. [8] Severe acne in these patients responds only to isotretinoin. [4]
Other reported cutaneous findings include hypopigmentation, hyperkeratosis, excessive skin wrinkling of the forehead, shoulders, elbows, and knuckles, [4] and oculocutaneous albinism. [5] In addition to severe acne, our patient also had generalized hypertrichosis especially as faun tail in the lumbosacral region and hyperhidrosis. He had also unilateral lymphedema with no defined cause.
Cleft palate or bifid uvula is found in approximately 75% of patients. Dental anomalies such as impacted teeth, delayed eruption, ectopic eruption, supernumerary teeth, and thick gingiva are also common. [4] The nose is down-turned at the tip, the bridge is depressed, and the septum is deviated. [5]
The usual hand deformity in AS consists of a bony fusion of the second, third, and fourth fingers. Involvement of the first or fifth digits in this bony mass is variable. There can be a similar deformity involving the foot (mitten hand and sock foot). Other skeletal abnormalities include limited mobility at glenohumeral joint and elbow joint, multiple epiphyseal dysplasia, very short or absent neck of scapula, small capitulum, and flat radial head. [5]
The ocular manifestations, hypertelorism and exophthalmia, seem to be present in all cases. The ocular orbits are shallow and the accompanying exophthalmia may lead to blindness. [4]
The most frequently reported cranial malformation is bilateral craniosynostosis of the coronal and lambdoid sutures along with reduction of the anterior, deep middle and posterior cranial fossae that results in displacement of the brain structures. [4]
Mental retardation occurs in most patients with AS, and may be due to brain malformations, or high intracranial pressure. [4],[7] Some reports have stated that many of AS patients have normal intelligence. [5] Our patient also had normal intelligence.
Other commonly associated systemic features include cardiac anomalies, and hearing defects. [5] Our patient was afflicted with PDA.
Acne in AS is often resistant to conventional therapies. [6],[8] Isotretinoin may be a valuable tool in the management of the severe acne afflicting these patients. [4] It is typically administered orally in doses of 0.5 to 2.0 mg/kg/day for a finite period of time (usually 16-20 weeks). Most case reports found that these patients benefit on a starting dose of 1 mg/kg/day; [4] our patient showed notable improvement with initiating dose of 0.5 mg/kg/day. Oral hormonal contraceptives affect acne in several ways in girls with AS. [6]
| 1. |
De D, Narang T, J Kanwar A, Dogra S. Brachycephaly and syndactyly: Apert's syndrome. Indian J Dermatol Venereol Leprol 2008;74:395-6.
[Google Scholar]
|
| 2. |
Verma SH, Draznin M. Apert syndrome. Dermatol Online J 2002;11:15.
[Google Scholar]
|
| 3. |
Dolenc-Voljc M, Finzgar-Perme M. Successful isotretinoin treatment of acne in a patient with Apert syndrome. Acta Derm Venereol 2008;88:534-5.
[Google Scholar]
|
| 4. |
Tiwari A, Agrawal A, Pratap A, Lakshmi R, Narad R. Apert syndrome with septum pellucidum agenesis. Singapore Med J 2007;48:62-5.
[Google Scholar]
|
| 5. |
Taksande A, Vilhekar K, Khangare S. Apert syndrome: a rare presentation. J Indian Acad Clinic Med 2007;8:245-6
[Google Scholar]
|
| 6. |
Hsieb T, Ho N. Resolution of acne following therapy with an oral contraceptive in a patient with Apert syndrome. J Am Acad Dermatol 2005;53:173-4.
[Google Scholar]
|
| 7. |
Mukhopadhyay AK, Mukherjee D. Apert's syndrome. Indian J Dermatol Venereol Leprol 2004;70:105-07.
[Google Scholar]
|
| 8. |
Benjamin LT, Trowers AB, Schachner LA. Successful acne management in Apert syndrome twins. Pediatr Dermatol 2005;22:561-5.
[Google Scholar]
|
Fulltext Views
3,590
PDF downloads
2,468





![[Figure - 1]](#fig_ijdvl_2010_76_6_724_72479_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2010_76_6_724_72479_f2.jpg){kind=link}