
Translate this page into:
Atypical adult type pityriasis rubra pilaris
Corresponding author: Dr. Pooja Dilip Shah, Department of Dermatology, Venereology & Leprosy, Gujarat Medical Education and Research Society Medical College & Hospital, Gandhinagar, India. poojarderm2020@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Shah PD, Padhiar BB, Sreelakshmi TS. Atypical adult type pityriasis rubra pilaris. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_1186_2024
Dear Editor,
Pityriasis rubra pilaris (PRP) is a rare idiopathic inflammatory papulosquamous disorder with bimodal age distribution (first and fifth decade), no sex predilection and has a chronic to self-limiting course which is clinically characterised by erythematous follicular papules coalescing to form orange-red plaques with islands of sparing, bran-like scaling and palmoplantar keratoderma (PPKD).1,2 Incidence of PRP in the Indian population is approximately 1 in 50,000 patients.3 Griffith’s classification distinguishing six types of PRP is most widely accepted. Herein, we report a case of Type II PRP which is an extremely uncommon variant with atypical clinical features.
A 27-year-old married Indian woman, born out of non-consanguineous marriage, presented with complaints of progressive thickening, scaling and deep fissures over her palm and soles for the last six months. This was followed by progressive generalised fish-like scaling over the body associated with itching and for the last 20 days she had begun to develop red colour skin lesions, which first appeared over her face followed by other flexural areas of the body. There was no history of collodion membrane at birth, drug intake or irritant application prior to the development of lesions, altered sweat production or similar complaints in her past and family. Cutaneous examination revealed diffuse erythema over the face with fine white scaling; bright red to orange scaly plaques on her neck, flexural areas, dorsa of hands and forearms; generalised coarse ichthyosiform scaling over the rest of her body; PPKD, subungual hyperkeratosis, nail dystrophy and inflamed nail folds with loss of cuticle [Figure 1a to 1d]. No abnormalities of mucosa, scalp or teeth were found. Systemic and laboratory evaluation was not significant. Dermoscopy showed white keratotic follicular plugging and furfuraceous scaling against the background of erythema [Figure 2a and 2b]. PRP, psoriasis, follicular eczema, ichthyosis, erythrokeratoderma were considered as differential diagnoses and a skin biopsy was performed from the erythematous scaly plaque on the extensor aspect of her right forearm. Histopathology revealed hyperkeratosis, orthokeratosis alternating with parakeratosis, follicular plugging, irregular acanthosis with broad rete ridges, thick suprapapillary plates and superficial perivascular lymphocytic infiltrate in the dermis [Figure 3a and 3b]. Based on clinicopathological correlation she was diagnosed with atypical adult (type 2) PRP and was started on oral isotretinoin 20 mg/day (which was gradually tapered following clearance of lesions over the course of six months), antihistamine (Tab. Levocetrizine 5mg BD), topical corticosteroids and emollients. She was monitored for six months after discontinuing treatment during which there was no recurrence.
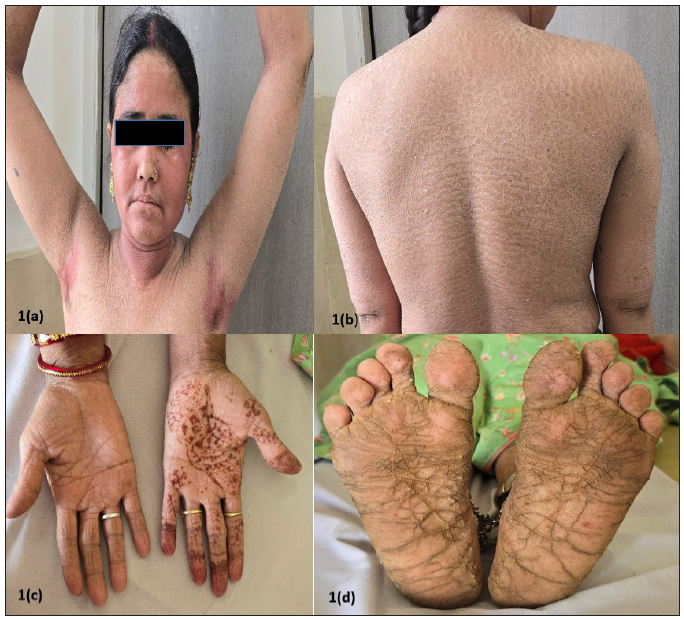
- (a) Diffuse erythema with fine scaling on the face and erythematous scaly plaques around the neck and axillae; (b) ichthyosiform scaling on the back; (c) and (d) Palmo-plantar keratoderma.
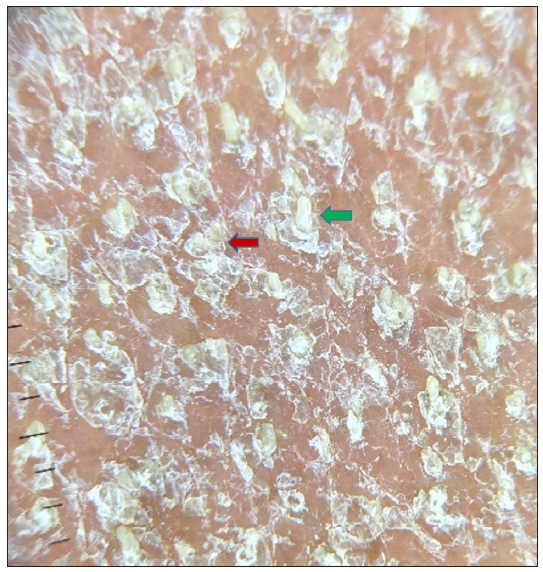
- Dermoscopy (using Illuco IDS-1100, 10x, polarised mode) shows white keratotic follicular plugging (green arrow) and furfuraceous scaling (red arrow) against the background of erythema (before treatment).

- Dermoscopy (using Illuco IDS-1100, 10x, polarised mode) shows white keratotic follicular plugging (green arrow) and furfuraceous scaling (red arrow) against the background of erythema (three weeks after treatment).
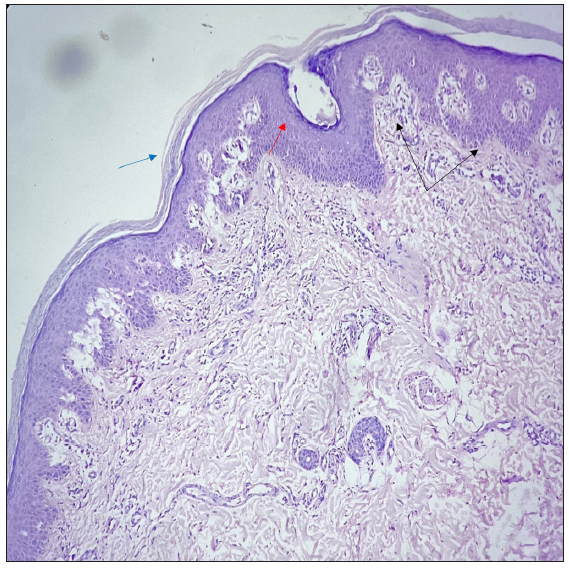
- Histopathology showing hyperkeratosis, orthokeratosis alternating with parakeratosis (blue arrow), follicular plugging (red arrow), irregular acanthosis with broad rete ridges, thick suprapapillary plates (black arrows), and superficial perivascular lymphocytic infiltrate in the dermis (Haematoxylin and eosin, 20x).
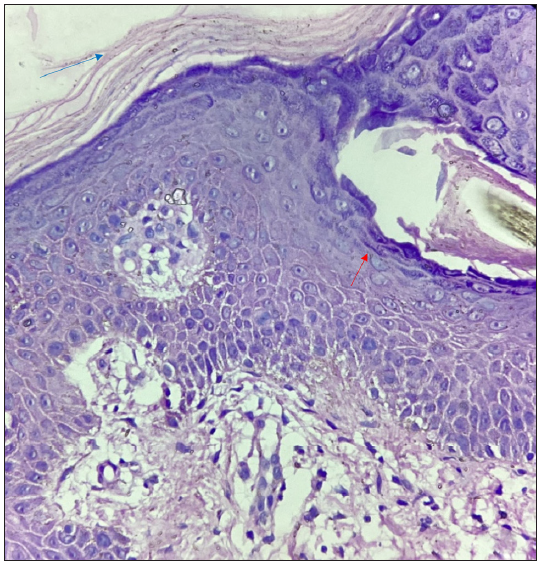
- Histopathology showing hyperkeratosis, orthokeratosis alternating with parakeratosis (blue arrow), and follicular plugging (red arrow) (Haematoxylin and eosin, 40x).
Mostly PRP is an acquired disorder but familial cases with autosomal dominant or recessive inheritance pattern involving mutations in CARD14/PSORS2 gene have been described.4
Various theories have been postulated as to its etiology, ranging from vitamin A deficiency, autoimmune etiology, an abnormal immune response to certain antigens, physical triggers like ultraviolet (UV) exposure, skin trauma, superantigens and drugs like imatinib, ponatinib and sofosbuvir.3,5,6
Classically, the disease progresses cephalocaudally with follicular hyperkeratosis being the elemental lesion; these follicular papules change from acuminate to flat-topped to be replaced by sheets of erythema (characteristic orange or yellowish hue) and pityriasiform/furfuraceous scaling with islands of sparing (nappes claires). Less commonly, the development of orange-red waxy PPKD (PRP sandal) may be the early presenting feature.1,7 Nails may show subungual hyperkeratosis, splinter haemorrhages, longitudinal ridging and thick discoloured nail plate. Though alopecia is uncommon, mucosal and eye lesions have been recorded.1
Atypical adult onset PRP afflicting about 5% of the patients was so named due to its chronic course (20 or more years) and presence of atypical clinical features like ichthyosiform scaling (especially on the legs), eczematous changes, sparseness of the scalp hair, coarse and lamellated palmoplantar hyperkeratosis. In his study, Griffith had mentioned that though these patients have no family history they might be having an ichthyosis-like disorder. The cephalocaudal progression is not seen and there are less chances of developing erythroderma. Only 20% of type II patients experience clinical resolution within three years.1,2
First-line therapy for PRP patients is generally oral retinoids (isotretinoin 1–1.5 mg/kg per day or acitretin 0.5–0.75 mg/kg/day) or methotrexate (weekly oral or subcutaneous in dose of 10–25 mg). Topical therapy with emollients, corticosteroids, vitamin D analogues and keratolytics act as an adjuvant. Other agents which have been used with variable results are azathioprine, cyclosporine, apremilast, TNF-α inhibitors, secukinumab, ustekinumab, alitretinoin, phototherapy and extracorporeal photopheresis.7
Interestingly, our patient had first developed PPKD followed by generalised ichthyosiform scaling, erythematous plaques predominantly involving flexures and dorsa of hands and forearms. Her face was red, taut and covered with fine powdery scales. Nails showed subungual hyperkeratosis, dystrophy and features of chronic paronychia. There was no cephalocaudal spread or sparseness of scalp hair. Last to resolve was facial erythema and PPKD while erythematous plaques and ichthyosiform scaling cleared up within 2–2.5 months of treatment. We wanted to elucidate this case due to the paucity of related literature.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Original article Pityriasis rubra pilaris‐an historical approach: 2 Clinical features. Clinical and experimental dermatology.. 1976;1:37-50.
- [CrossRef] [PubMed] [Google Scholar]
- Pityriasis rubra pilaris. Clinical and experimental dermatology.. 1980;5:105-12.
- [CrossRef] [PubMed] [Google Scholar]
- Other papulosquamous disorders. In: Dermatology Vol Vol. 1. (4th ed). Elsevier; 2017. p. :166-170.
- [Google Scholar]
- Familial pityriasis rubra pilaris is caused by mutations in CARD14. The American Journal of Human Genetics.. 2012;91:163-70.
- [CrossRef] [Google Scholar]
- Pityriasis rubra pilaris and human immunodeficiency virus infection. British Journal of Dermatology.. 1995;133:990-93.
- [CrossRef] [Google Scholar]
- Ichthyosiform pityriasis rubra pilaris-like eruption secondary to ponatinib therapy: case report and literature review. Drug safety-case reports.. 2017;4:1-4.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Fitzpatrick’s Dermatology (9th ed). New York: McGraw-Hill Education; 2019. p. :498-504. Chapter 29, Pityriasis rubra pilaris




