Translate this page into:
Atypical dyskeratosis congenita
Correspondence Address:
M Jayaraman
222 RK Mutt Road, Mylapore, Madras-600004.
India
| How to cite this article: Krishnan SS, Yesudian P D, Jayaraman M, Janaki V R, Raj JB. Atypical dyskeratosis congenita. Indian J Dermatol Venereol Leprol 1997;63:47-49 |
Abstract
Dyskeratosis congenita is a syndrome characterised mainly by pigmentation and atrophy of skin, nail dystrophy and oral leucoplakia. We report a patient who had features consistent with this syndrome including skin atrophy and pigmentation, oral leucoplakia, oesophageal stricture, but with normal finger and toe nails. Even though many variants have been described in the literature sparing of the nails as in our patient is extremely uncommon.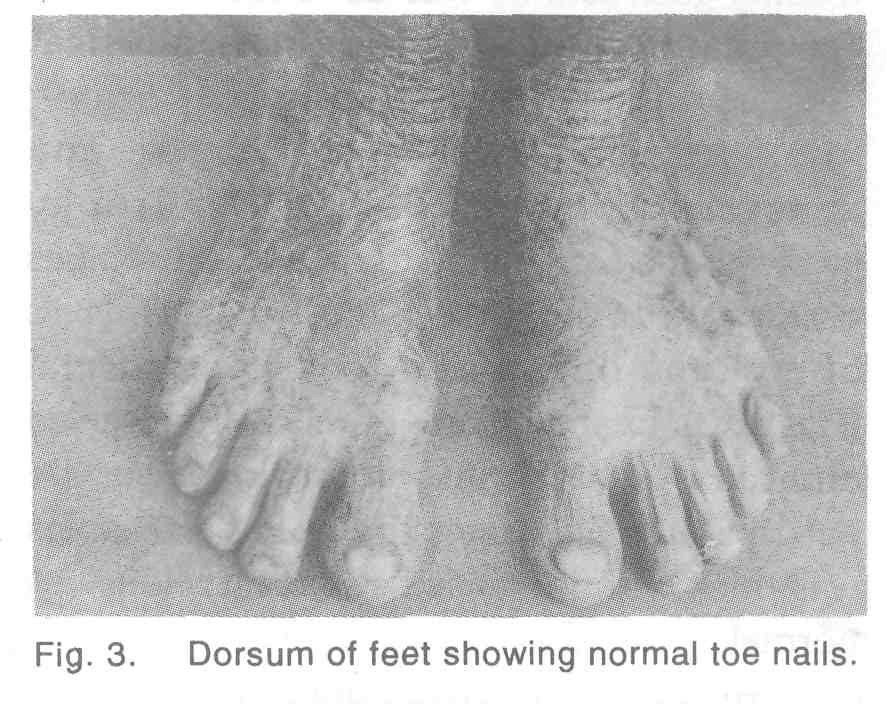 |
|
 |
|
 |
|
Introduction
Cellular DNA can be damaged by irradiation or mutagenic or carcinogenic chemicals in the environment. Normal individuals possess the ability to repair this damage by a complex series of biochemical pathways. In some diseases, ability to repair damage to the genome is impaired, with features of chromosomal instability and an increased risk of malignancy.[1] These include xeroderma pigmentosum, Bloom′s syndrome, Cockayne′s syndrome, dyskeratosis congenita and progeria. The following case report is an atypical presentation of dyskeratosis congenita. This is a genetically determined disorder transmitted in a X-linked recessive fashion, though autosomal dominant inheritance has also been reported.[2] The essential features of this syndrome are atrophy and pigmentation of skin, nail dystrophy and oral leucoplakia.
Case Report
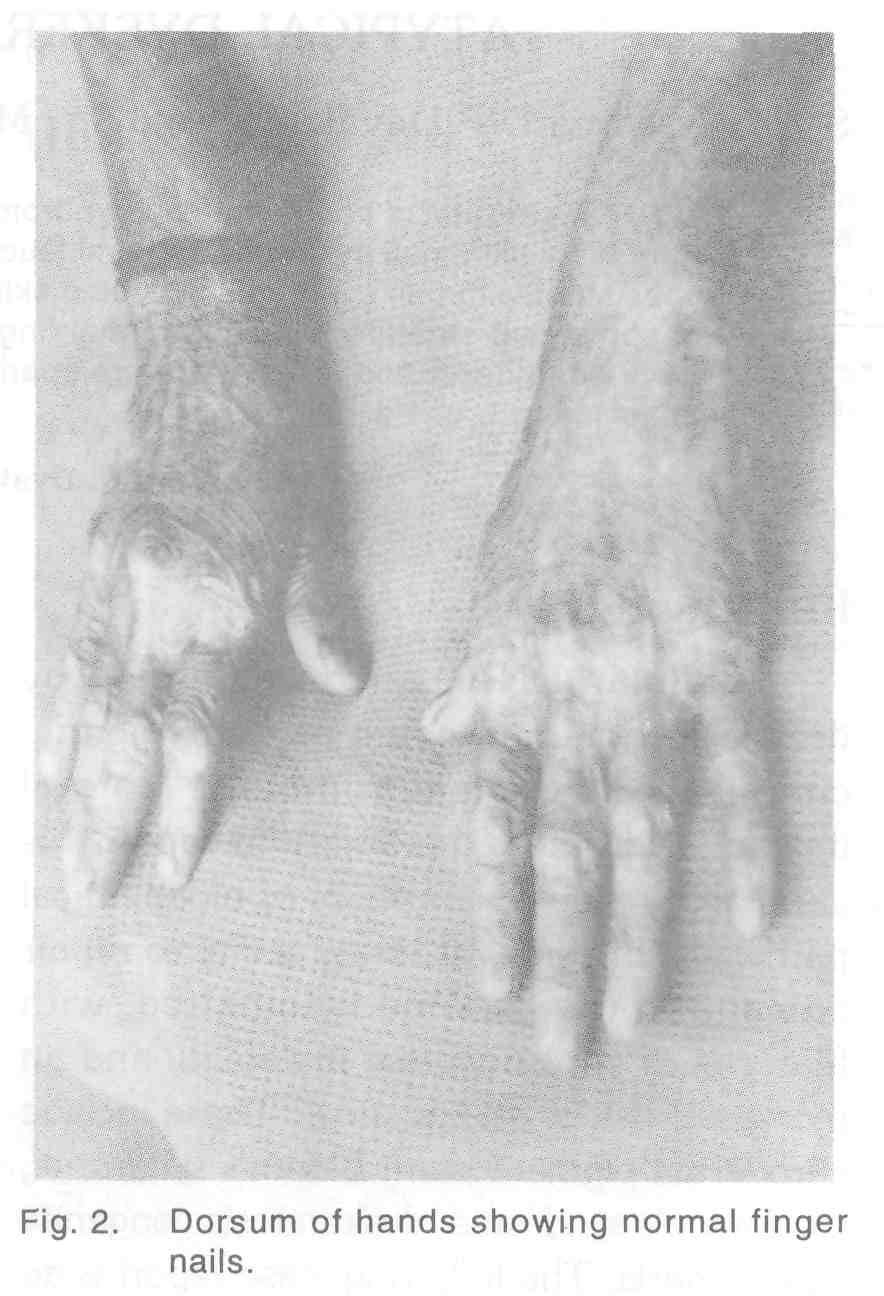
A 24-year-old man was referred to the dermatology OP from Gastroenterology department as a case of scleroderma with difficulty in swallowing. The patient was the second child born to non-consanguineous parents and was normal at birth. From the age of 6 years, he developed erythema of the skin with blisters on exposure to sunlight. These had healed with areas of pigmentation and depigmenation. Patient denied developing any blisters over the oral cavity. At the age of 18 years, he developed pain and swelling over the gums. At that time it was found that most of his teeth were carious. Dental extraction was done and patient was given dentures. Now for that past 1 year patient has difficulty in swallowing which was diagnosed as stricture oesophagus and a feeding gastrostomy was done for the same. On examination mental and physical development of the patient was normal. There were areas of hypo and hyperpigmentation of skin with atrophy involving the face, neck, trunk and extremities giving a picture of poikiloderma [Figure - 1]. Nails of both the fingers and toes were normal [Figure - 2],[Figure - 3]. Examination of oral cavity revealed an area of whitish discolouration over the hard palate. Skin over the palms and soles was thick with increased sweating. Hair was normal.
Blood investigations did not reveal any anemia or pancytopenia. Readiography of the skull did not show any area of calcification. A biopsy from the area of poikiloderma showed atrophy of epidermis with macrophages in the dermis laden with melanin. Basal cell degeneration was not prominent.[3] A diagnosis of dyskeratosis congenita was made and patient was given sun screens and advised to avoid sunlight and asked to come regularly for follow up.
Discussion
Dyskeratosis congenita (Zinsser-Cole-Engman syndrome) was first described in 1960 by Zinsser.[4] It is characterised by atrophy and a reticular pigmentation of the skin, dystrophy of the nails and mucosalleuko keratosis, together with multi system ectodermal and mesodermal changes.[5]
Generally nail changes are the first component of the syndrome to appear. Nails become dystrophic with thinning, tapering and they may be reduced to horny plugs or be completely destroyed. In mild cases ridging and longitudinal fissuring occur. In our patient all the toe and finger nails were normal with no history of being shed in the past.
Involvement of oral cavity with leukoplakia, periodontal disease with premature caries of teeth, stricture of oesophagus and other areas of GIT along with pigmentary changes of skin as seen in our patient is commonly seen in persons with this condition. There were no hematological or skeletal abnormalities or malignancies or involvement of the genitourinary tract in our patient although they have been reported in association with dyskeratosis congenita.[1]
Nail involvement is a common and prominent finding in any case of dyskeratosis congenita. To the best of our knowledge, the occurrence without nail chagnes is very rare, though other component may be absent in partial forms. This emphasizes the fact that not all component of the syndrome need to be present in any given patients and in any genodermatosis, pleomorphism may be expected with variable clinical manifestations of a single genetic aberration.
| 1. |
Harper J. Genetics and genodermatoses. In : Champion RH, Burton JL, Ebling FJG, eds. Textbook of dermatology. 5th edn. Oxford : Blackwell, 1992:54-6.
[Google Scholar]
|
| 2. |
Tchou PK, Kohn T. Dyskeratosis congenita : an autosomal dominant disorder. J Am Acad Dermatol 1982;6:1034-9.
[Google Scholar]
|
| 3. |
Bryan HG, Nixon RK. Dyskeratosis congenita and familial pancytopenia. JAMA 1965;192:203-8.
[Google Scholar]
|
| 4. |
Zinsser F. Atrophia cutis reticularis cum pigmentione, dystrophia unguium et leukokeratosis oris. Ikonogr Dermatol 1960;5:219-23.
[Google Scholar]
|
| 5. |
Arnold HL, Odom RB, James WD. Some genodermatosis. In : Arnold HL, Odom RB, James WD, eds. Andrew's diseases of the skin. 8th edn. Philadelphia : WB Saunders, 1990:674.
[Google Scholar]
|





![[Figure - 1]](#fig_ijdvl_1997_63_1_47_4505_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_1997_63_1_47_4505_2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_1997_63_1_47_4505_3.jpg){kind=link}