Translate this page into:
Bardet-Biedl syndrome: A rare case report from North India
Correspondence Address:
Jyotisterna Mittal
102, Good Earth Colony, B/s New Moti Palace, Patiala - 147 001, Punjab
India
| How to cite this article: Kumar S, Mahajan BB, Mittal J. Bardet-Biedl syndrome: A rare case report from North India. Indian J Dermatol Venereol Leprol 2012;78:228 |
Abstract
The Bardet-Biedl syndrome (BBS) is a rare ciliopathic human autosomal-recessive disorder, affecting multiple organ systems. Less than 15 cases have been reported from India. The authors present a classical case of BBS presenting to dermatology outpatient with hypogonadism and features such as marked central obesity, retinal dystrophy, polydactyly, structural renal abnormalities and mental retardation, along with a brief review of the literature. This case exemplifies the need for multidisciplinary management in such cases.Introduction
The Bardet-Biedl syndrome (BBS) is a rare ciliopathic human autosomal-recessive disorder, which is characterized principally by cardinal symptoms of marked central obesity, retinal dystrophy, polydactyly, mental retardation and hypogonadism and renal dysfunction. [1] The frequency of the syndrome is estimated to be 1:1,60,000. [2] Less than 15 cases have been reported from India. [3] The incidence is much higher in some populations with a high level of consanguinity or those that are geographically isolated, with disease incidence of 1 in 13,000 in the isolated populations of Newfoundland and Kuwait, 1 in 17,000 live births. [4] The authors present a classical case of BBS presenting to the dermatology outpatient with hypogenitalism and features such as marked central obesity, retinal dystrophy, polydactyly, structural renal abnormalities and mental retardation along with a brief review of the literature.
Case Report
A 14-year-old boy reported to the skin outpatient department with complaint of underdevelopment of genitalia and absence of hair on the pubic and axillary areas [Figure - 1]. His background problems included behavioral issues and diminished vision in both eyes, especially at night. He was a product of consanguineous marriage. There is history of death of one of the older male siblings at 18 months of age reportedly due to unknown reasons and death of a maternal cousin, who had similar phenotypic characters, at 11 years of age due to nephropathy. The initial motor and developmental milestones of our case were delayed, with commencement of walking and speech at 3 and 5 years of age, respectively.
 |
| Figure 1: The 14-year-old boy with central obesity, underdeveloped genitalia and absence of hair on the pubic and axillary areas |
Mental development was lagging behind the normal range, with an I.Q. less than 70. Height and weight were 130 cm and 65 kg, respectively, with a resultant body mass index of 38, which indicates severe obesity. Blood pressure and pulse rate were 110/70 mm of Hg and 84 per minute, respectively. There was proximal lower limb weakness (power grading of 4 out of 5). The plantar reflex was exaggerated. There was postaxial polydactyly with hexadactyly of all four limbs [Figure - 2] and [Figure - 3] and features of hypogonadism with testicular volume of 2 ml (normal 10-12 ml) and micropenis (<2.5 cm) [Figure - 4]. Magnetic resonance imaging of the brain was performed, and revealed normal study.
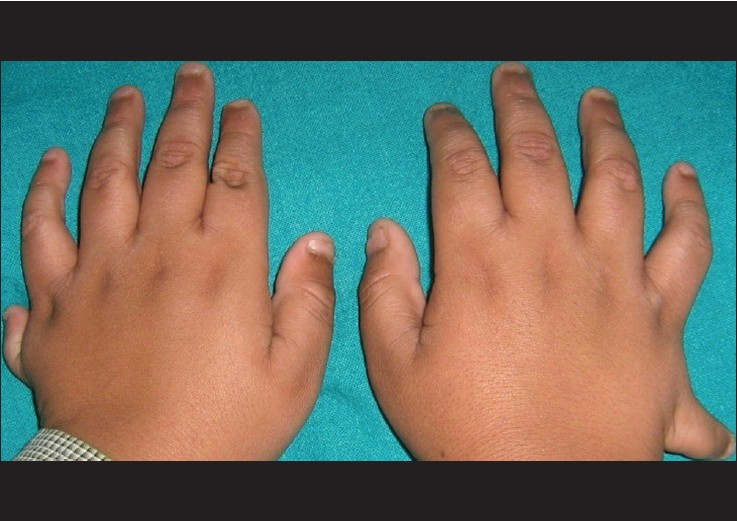 |
| Figure 2: Postaxial polydactyly with hexadactyly of hands and feet |
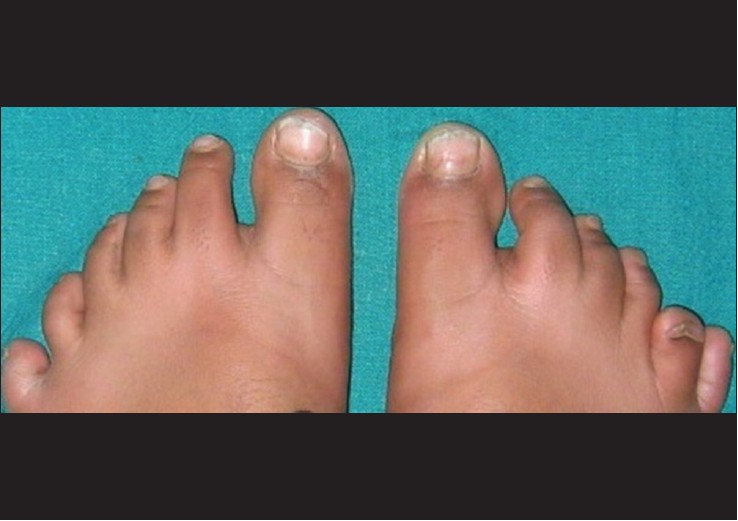 |
| Figure 3: Postaxial polydactyly with hexadactyly of hands and feet |
 |
| Figure 4: Hypogonadism with micropenis and absence of secondary sexual characters |
On ophthalmological examination, visual acuity was 6/36 in both eyes. Fundus examination showed pallor of the optic disc, bilaterally attenuated vessels and retinal pigmentary changes of retinitis pigmentosa "sine pigmento" variety [Figure - 5], with mild night blindness and constricted visual field. The child also had bilateral conductive deafness of moderate degree.
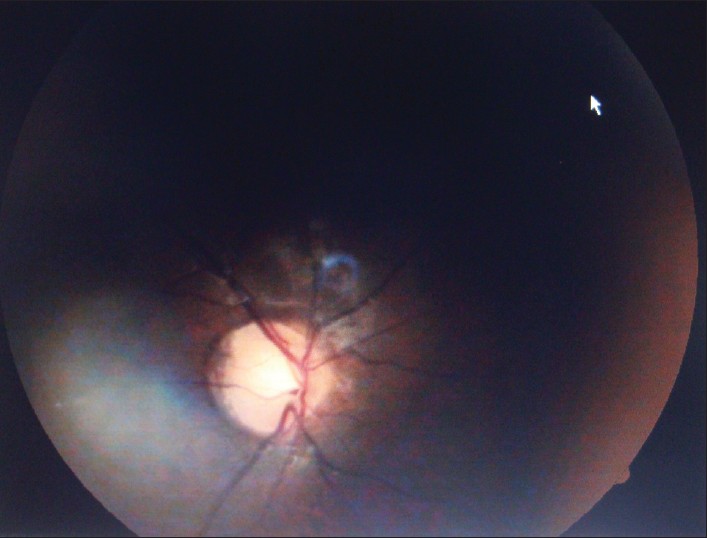 |
| Figure 5: Fundus photography with features of retinitis pigmentosa sine pigmento |
Apart from deranged liver function tests, other investigations like hemogram, renal function tests and urine microscopy were normal. Ultrasound revealed fatty liver and increased cortigenicity of renal cortex along with irregular margins. Electrocardiogram and echocardiogram were done to rule out underlying cardiac abnormalities, and were normal.
Parents of the child were counseled and called for regular follow-up to observe whether any progressive renal changes develop and for behavioral therapy. The patient was also put on testosterone supplementation as per endocrinological consultation.
Discussion
BBS is named after Georges Bardet and Arthur Biedl. The first known case was reported by Laurence and Moon in 1866. Laurence-Moon-Biedl-Bardet syndrome (LMBBS) is no longer considered as a valid term as patients of Laurence and Moon had paraplegia but no polydactyly and obesity, which are the key elements of the BBS. Hence, Laurence-Moon syndrome is usually considered a separate entity. However, some recent research suggests that the two conditions may not be distinct. [4]
Apart from the cardinal manifestations, other features of the BBS include various degrees of intellectual impairment, congenital heart block, brachycephaly, deafness and dental anomalies. The full spectrum of clinical features is found in only 40-45% of LMBBS cases. [5] Hypogonadism, which is probably primary in origin, is reported more frequently in males than in females. [6]
The detailed biochemical mechanism that leads to BBS is still unclear. Twelve genes (BBS1 to BBS12) that are responsible for the disease have been cloned. The BBS proteins are components of the centrosome and affect the ciliary transport; hence, the disease falls under the spectrum of "ciliopathies." [7]
Puberty is a particularly stressful time for those with BBS, and referring the patient to an experienced counselor is especially helpful. testosterone supplements may be prescribed to male patients, specifically in cases having lowered level of this hormone. There are no proven effective treatments to either prevent or alleviate the deterioration in vision. However, spectacles were advised as low vision was present in our patient, and regular ophthalmological follow-up was stressed upon. Accessory digits are often nonfunctional and may be excised. Obesity is an area of major concern in these patients and, if left uncontrolled, will lead to multiple health problems. A low-calorie and low-protein diet help in obesity control and may slow the progression of renal failure in patients with BBS. [8] The patient was advised diet control as per the dietician′s recommendations, and physical exercises.
As structural renal changes were identified in our patient, he was advised half-yearly urinalysis, blood pressure and urea and creatinine levels. Any derangement in these renal function tests would warrant prompt referral to a nephrologist as end-stage renal disease is an important cause of morbidity, especially in pediatric patients. [3]
In general, BBS patients are happy and friendly children, and the society should not have a discriminatory attitude toward these children so that their quality of life may be improved.
Our patient presented with the classical phenotype of BBS. Even though the patient had been seen by several specialists in different facilities, the diagnosis had been missed, possibly because of the rarity of the condition. It was the hypogonadism and the absence of secondary sexual characters that brought the patient to our department, where the diagnosis was made. The case is being reported for its rarity.
| 1. |
Beales PL, Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J Med Genet 1999;36:437-46.
[Google Scholar]
|
| 2. |
Klein D, Ammann F. The syndrome of Laurence-Moon- Bardet-Biedl and allied diseases in Switzerland. Clinical, genetic and epidemiological studies. J Neurol Sci 1969;9:479-513.
[Google Scholar]
|
| 3. |
Hooda AK, Karan SC, Bishnoi JS, Nandwani A, Sinha T. Renal transplant in a child with Bardet-Biedl syndrome: A rare cause of end-stage renal disease. Indian J Nephrol 2009;19:112-4.
[Google Scholar]
|
| 4. |
Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, et al. Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: A 22-year prospective, population-based, cohort study. Am J Med Genet 2005;132:352-6.
[Google Scholar]
|
| 5. |
Prosperi L, Cordella M, Bernasconi S. Electroretinography and diagnosis of the Laurence-Moon-Bardet-Biedl syndrome in childhood. J Pediatr Ophthalmol 1977;14:305-8.
[Google Scholar]
|
| 6. |
Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, et al. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med 1989;321:1002-9.
[Google Scholar]
|
| 7. |
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranan J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 2007;129:1201-13.
[Google Scholar]
|
| 8. |
Dervisoglu E, Isgoren S, Kasgari D, Demir H, Yilmaz A. Obesity control and low protein diet preserve or even improve renal functions in Bardet-Biedl syndrome: A report of two cases. Med Sci Monit 2011;17:12-4.
[Google Scholar]
|
Fulltext Views
4,482
PDF downloads
1,660





![[Figure - 1]](#fig_ijdvl_2012_78_2_228_93656_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_2_228_93656_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_2_228_93656_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2012_78_2_228_93656_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2012_78_2_228_93656_f5.jpg){kind=link}