Translate this page into:
Bone marrow transplantation improves symptoms of congenital erythropoietic porphyria even when done post puberty
2 Department of Medical Oncology, All India Institute of Medical Sciences (AIIMS), New Delhi, India
Correspondence Address:
Neena Khanna
Department of Dermatology and Venereology, All India Institute of Medical Sciences, Ansari Nagar, New Delhi - 110 029
India
| How to cite this article: Singh S, Khanna N, Kumar L. Bone marrow transplantation improves symptoms of congenital erythropoietic porphyria even when done post puberty. Indian J Dermatol Venereol Leprol 2012;78:108-111 |
Sir,
Porphyrias are a group of rare yet interesting, usually hereditary disorders characterized by a whole gamut of cutaneous manifestations. It is mainly the cutaneous porphyrias that present primarily to the dermatologist. Among the group of cutaneous porphyrias that include congenital erythropoietic porphyria (CEP), porphyria cutanea tarda, erythropoietic protoporphyria and hepatoerythropoietic porphyria, it is CEP that is associated with the most dramatic clinical profile. Currently, there is no curative treatment for CEP except stem cell transplantation. [1] We report a post pubertal male with CEP who improved significantly after allogenic bone marrow transplantation.
A 14-year-old boy presented with photosensitivity, blistering on sun-exposed sites, facial hypertrichosis, acral mutilation and history of passing red-colored urine since birth. He was born out of nonconsanguineous marriage and had a younger healthy sibling. There were no systemic complaints other than easy fatiguability. Dermatological examination revealed facial hypertrichosis, erosions and irregular depressed scars and numerous eroded blisters on the acral parts with marked resorption of digits [Figure - 1]a and b.Erythrodontia was also evident in all the teeth, with fluorescence under Wood′s lamp. Hemogram showed moderate normocytic, normochromic anemia (hemoglobin-10 g/dl) with blood chemistry being within normal limits. Skin biopsy from a fresh blister revealed a pauci-inflammatory subepidermal blister with deposition of PAS-positive material around the papillary dermal blood vessels. The findings were compatible with a clinical diagnosis of porphyria. A qualitative analysis for porphyrins in blood, urine and stool under Wood′s lamp was positive compared to the corresponding samples from a healthy control. Because of the major impact of disease on patient′s quality of life, in that he had to remain indoors during the day because of marked photosensitivity and also because his anemia had not responded to hematinics and multiple blood transfusions given previously, allogenic bone marrow transplantation (BMT) from his human leukocyte antigen (HLA) matched 4-year-old male sibling was performed. Before harvesting the marrow stem cells, the donor was given granulocyte colony stimulating factor (G-CSF) (5 μg/kg subcutaneously twice a day for 5 days). Considering the risks of immunosuppression and graft versus host disease (GVHD), a BMT using a reduced intensity conditioning (low-dose cyclophosphamide-based regimen) was performed using cyclophosphamide (20 mg/kg divided on days -5 and -4), fludarabine (25 mg/m 2 / day on days -14 to -10) and anti thymocyte globulin (40 mg/kg/day on days -9 to -6) as part of the myeloablative regimen. Cyclosporine (5 mg/kg/day, initially intravenous [IV] and shifted to oral after successful engraftment) along with methotrexate (4 mg/m 2 IV for 5 days) was given for GVHD prophylaxis, with cyclosporine eventually tapered over 6 months and finally stopped. At 2 years and 4 years after BMT [Figure - 2]a and b, the patient had marked reduction in photosensitivity, absence of blistering, decreased redness of urine and decreased positivity of blood, urine and stool on Wood′s lamp examination [Figure - 3]a and b, with the hemogram being normal (Hb = 13.5 gm%) and absence of malaise or fatigability. The patient did not experience features of GVHD or side-effects to immunosuppressive drugs at any point during the follow-up. More importantly, the patient and his family were satisfied with the treatment as he was able to spend more time outdoors due to improved sun tolerance.
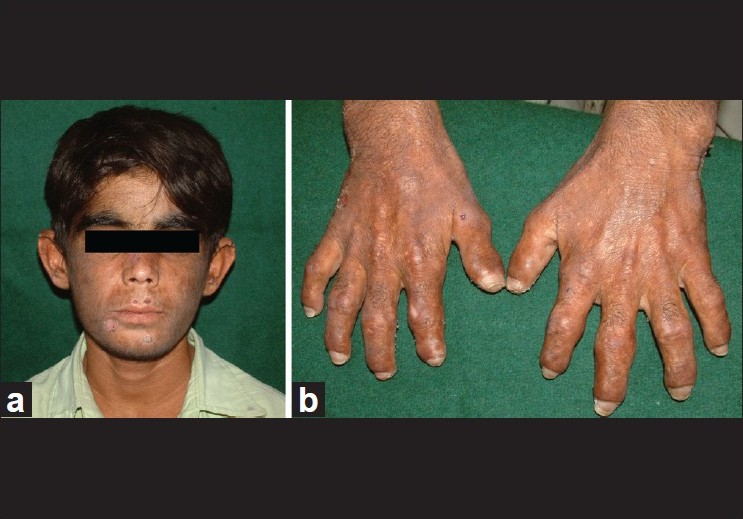 |
| Figure 1: Patient before bone marrow transplantation. (a) Facial hypertrichosis and healing blisters. (b) Acral mutilations |
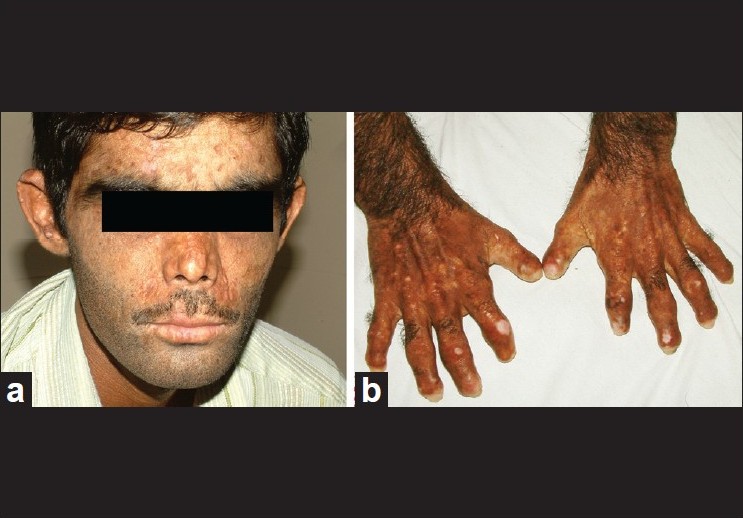 |
| Figure 2: Patient 4 years after bone marrow transplantation. (a) Significant reduction in photosensitivity and blistering. (b) Nonprogression of mutilation |
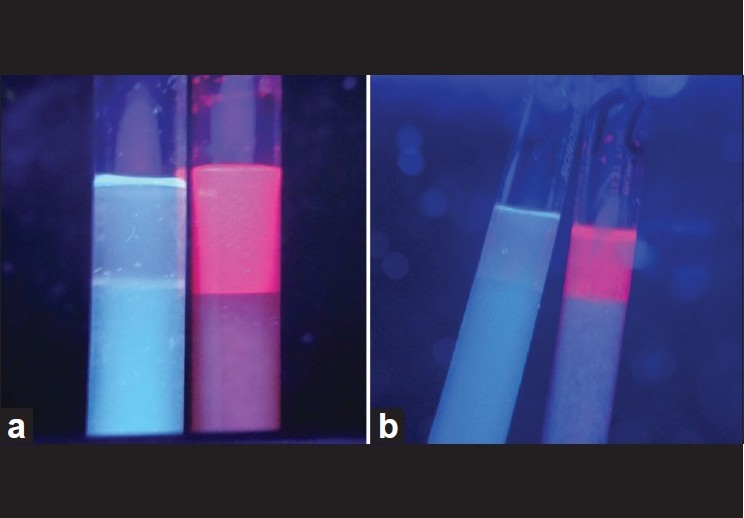 |
| Figure 3: (a) Wood's lamp examination of urine before bone marrow transplantation showing bright pink fluorescence in the patient's sample, with the control sample on the left side being negative. (b) Wood's lamp examination of urine 4 years after therapy, showing reduced positivity for porphyrins (lighter pink color) as compared with baseline [Figure 3a] |
CEP is a rare cutaneous porphyria caused by deficiency of uroporphyrinogen III synthase (URO III synthase), which has a variable clinical phenotype, ranging from severe lifelong photosensitivity, skin fragility, transfusion-dependent hemolytic anemia and hypersplenism with curtailed life expectancy, to only mild cutaneous features in some cases. [1]
Several treatment modalities have been described for CEP, including splenectomy, packed red blood cell transfusion and oral activated charcoal, but none of them is curative. [1],[2]
Stem cell transplantation has emerged as an effective treatment option for CEP because replacement of bone marrow erythroblasts corrects the erythroid defect. Engraftment of donor stem cells normalizes URO III synthase activity, decreases porphyrin levels, skin fragility and photosensitivity and re-establishes normal erythropoiesis. [3]
Kauffman et al.[4] first described the use of BMT for CEP in 1991 and, although their patient died of CMV pneumonitis 10 months after BMT, she had good improvement in her skin lesions. Since then, 12 more patients have undergone the procedure without mortality and with good clinical and biochemical remission of the disease. [5]
As far as ascertained in the English literature, our case is the oldest to undergo BMT for CEP at the age of 14 years and with post transplantation follow-up of 4 years. Farachi et al.[6] described the case of a 12-year-old boy who underwent hematopoietic stem cell transplantation from an unrelated volunteer donor, and the patient had persistently good improvement 7 years post transplant in terms of complete healing of skin lesions and absence of blistering on sun exposure, although teeth hyperpigmentation persisted.
Our case is also remarkable in that he tolerated immunosuppressive therapy well, and is the only case in the literature to have undergone BMT for porphyria using a low-dose cyclophosphamide-based regimen before the transplantation and yet having successful engraftment. In addition, he is also the only documented Indian patient of CEP to have undergone BMT.
The clinical benefit in our case (in terms of reduction in blistering, photosensitivity and mutilation) was not as drastic as has been achieved in other cases [3],[5],[6],[7] because the BMT was done very late thus highlighting the importance of early stem cell transplantation, which is curative.
Before BMT is performed for CEP, it requires careful and thorough evaluation, including clinical evaluation, quality of life and psychosocial factors, as also quantitative porphyrin estimation, which parallels disease severity, [8] and discussing the risks and benefits of the procedure with the patients and their families. Although porphyrin estimation could not be performed in our case, the clinical and psychosocial improvement without any therapy-related adverse events and the fact that he did not require hematinics or blood transfusions for anemia any further is proof enough for the safety and efficacy of BMT in post pubertal CEP patients.
Acknowledgement: We thank Mr Sushil Pandey, AIIMS, for helping with the qualitative porphyrin estimation.
| 1. |
Taibjee SM, Stevenson OE, Abdullah A, Tan CY, Darbyshire P, Moss C, et al. Allogeneic bone marrow transplantation in a 7-year-old girl with congenital erythropoietic porphyria: A treatment dilemma. Br J Dermatol 2007;156:567-71.
[Google Scholar]
|
| 2. |
Bicker DR, Frank J. The porphyrias. In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller BS, Leffell DJ, editors. Fitzpatrick's Dermatology in General Medicine. 7 th ed. New York: McGraw-Hill; 2008. p. 1228-56.
th ed. New York: McGraw-Hill; 2008. p. 1228-56. '>[Google Scholar]
|
| 3. |
Harada FA, Shwayder TA, Desnick RJ, Lim HW. Treatment of severe congenital erythropoietic porphyria by bone marrow transplantation. J Am Acad Dermatol 2001;45:279-82.
[Google Scholar]
|
| 4. |
Kauffman L, Evans DI, Stevens RF, Weinkove C. Bone-marrow transplantation for congenital erythropoietic porphyria. Lancet 1991;337:1510.
[Google Scholar]
|
| 5. |
Lebreuilly-Sohyer I, Morice A, Acher A, Dompmartin A, Clement C, de Verneuil H, et al. Congenital erythropoeietic porphyria treated by haematopoietic stem cell allograft. Ann Dermatol Venereol 2010;137:635-9.
[Google Scholar]
|
| 6. |
Farachi M, Morreale G, Boeri E, Lanino E, Dallorso S, Dini G, et al. Unrelated HSCT in an adolescent affected by congenital erythropoietic porphyria. Pediatr Transplant 2008;12:117-20.
[Google Scholar]
|
| 7. |
Tezcan I, Xu W, Gurgey A, Tuncer M, Cetin M, Oner C, et al. Congenital erythropoietic porphyria successfully treated by allogenic bone marrow transplantation. Blood 1998;92:4053-8.
[Google Scholar]
|
| 8. |
Freesemann AG, Bhutani LK, Jacob K, Doss MO. Interdependence between degree of porphyrin excess and disease severity in congenital erythropoietic porphyria (Gunther's Disease). Arch Dermatol Res 1997;289:272-6.
[Google Scholar]
|
Fulltext Views
3,533
PDF downloads
3,277





![[Figure - 1]](#fig_ijdvl_2012_78_1_108_90963_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_1_108_90963_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_1_108_90963_f3.jpg){kind=link}