
Translate this page into:
Clinical and molecular studies in two patients with dystrophic epidermolysis bullosa
 , Manoj Srinivasa1, Sheetal Desai1, R Mala1, Charitha K Jayashankar1, Rai Abhigna, Vishwanth Jyothi, Kubba Asha, Batrani Meenakshi2, Hiremagalore Ravi1, Baraka Vishwanthan Gurudatta
, Manoj Srinivasa1, Sheetal Desai1, R Mala1, Charitha K Jayashankar1, Rai Abhigna, Vishwanth Jyothi, Kubba Asha, Batrani Meenakshi2, Hiremagalore Ravi1, Baraka Vishwanthan Gurudatta
-
Received: ,
Accepted: ,
How to cite this article: Ramesh A, Hongal A, Srinivasa M, Desai S, Mala R, Jayashankar CK, et al. Clinical and molecular studies in two patients with dystrophic epidermolysis bullosa. Indian J Dermatol Venereol Leprol 2023;89:880-3.
Sir,
Epidermolysis bullosa is a group of rare genetic mechano-bullous diseases of the skin resulting in early blistering of the skin and mucous membrane. Bart syndrome or type VI aplasia cutis congenita is characterised by the clinical triad of congenital, localised absence of skin on limbs, mucocutaneous blistering (any type of epidermolysis bullosa) and nail abnormalities.1–3 This paper describes the clinical features, molecular studies of two babies with dystrophic EB congenita including exome sequencing and the care provided to them.
A three-days-old female baby was brought with peeling of skin associated with fluid-filled lesions on lower extremities, trunk and absence of skin on certain areas of both lower limbs since birth. There was no positive family history. A thin shiny translucent membrane was seen on the anteromedial aspect of both lower limbs and feet, consistent with aplasia cutis congenital (Figure 1 Patient 1 newborn). There was no mucosal involvement, but perioral desquamation and erosions were present. Systemic examination was normal. Paraffin-based dressings were advised. During the follow-up at 18 months of age, previously eroded areas as well as the aplastic areas had developed atrophic scarring. There was onycho-dystrophy of both thumbs, great toes, right fourth toe and anonychia of left third, fourth fingernails and all except the great toe nail on the right (Figure 1 patient 1-one year). The second baby, a five-days-old male, presented with a congenital absence of skin over the right lower limb (Figure 1 Patient 1 newborn). He was born to third-degree consanguineous parents [Figure 2b]. The paternal uncle had similar lesions with blister formation since birth and died at 25 years of age owing to complications arising out of his skin problems. Physical examination showed an asymmetrical absence of skin over the anterior-medial aspect of both legs [Figure 1b] (Figure 1 Patient 2) and a few blisters over the dorsa of fingers. The wounds were treated with a gentle two-sided wound contact layer with silicone adhesion (MepitelTM) and collagen-based dressings. The healed wounds showed hypopigmentation, milia and minimal atrophy. Immunofluorescence mapping IFM on skin biopsy of the second baby did not reveal any detectable staining of type VII collagen and showed reduced type VII collagen staining compared to control skin, and normal staining was seen for type IV collagen, laminin 332 and keratin 14 antibodies. These findings were suggestive of dystrophic epidermolysis bullosa [Figure 2a]. Genetic testing (exome sequencing) of the first baby identified a de novo heterozygous missense mutation nucleotide change in c.7976G>A; p. Gly2659Glu in the collagen VIIa triple helix domain with parents harbouring normal alleles [Figure 2b-c]. In silico analysis with polyphen and sift tools, and predictions suggested that the variant is pathogenic and deleterious [Figure 2d]. Exome analysis of second patient identified two homozygous mutations in exons 19 and 85 in the collagen VIIa gene –mutation with nucleotide changes c.2548G>T p. Asp850Tyr located in fibronectin repeat domain (Figure 2d-patient 1) and c.6738G>T p. Leu2246Phe in the triple helix domain, respectively (Figure 2d-patient 2) These changes are predicted to be deleterious and reduce the stability of the protein (Figure 2d-patient 2). Both above variants were novel and has not been previously described in reference databases. Correlation of clinical and IFM findings along with molecular genetic analysis of probands have identified both dominant dystrophic epidermolysis bullosa (case1) and recessive dystrophic epidermolysis bullosa (case 2) in association with aplasia cutis congenita (class VI) [Figure 2b]. These findings are summarised in Table 1.
Several studies have identified missense mutations in the triple helix domain of collagen type VII alpha 1 chain with epidermolysis bullosa and aplasia cutis congenita [Figure 3]. These mutations in the triple helix domain can destabilise the triple helix, ultimately leading to the formation of abnormal anchoring fibril complex and skin fragility. This was reflected in the reduced collagen VIIa staining in the dermis which suggests that mutant protein could be targeted for ubiquitin-mediated degradation. On the other hand, mutations in NC1 (case 2) could affect the domain interactions with critical partners in the extra cellular matrix, such as binding to fibronectin. Repeated blistering at mechanically stressed areas and altered healing lead to persistent inflammation, which often manifests as frequent infections, fibrosis, scarring and deformity.4 , 5 For a dermatologist, with limited treatment options, accurate diagnosis and counselling are the key steps in devising a treatment plan. Wound care with supplemental collagen-based dressing is critical for the early management of dystrophic epidermolysis bullosa. This is essential to check the progression of the wounds into scars and deformities in multidisciplinary care. Information obtained from genetic testing would be prudent in deploying cascade screening, prenatal testing and genetic counselling for future pregnancies.
| Inheritance | Clinical manifestation | IFM | Domain of protein | PolyPhen2.0/SIFT | |
|---|---|---|---|---|---|
| Patient 1 | Autosomal dominant (de novo) | Absent skin was seen as a thin shiny translucent membrane on the anteromedial aspect of both lower limbs and the feet, consistent with aplasia cutis congenital Onycho-dystrophy of bilateral thumb, great toes and right 4th toe, and anonychia of left 3rd, 4th fingernails and right 2nd to 5th toenails. | Absent staining of type VII collagen | p. Gly2659Glu triple helix domain (THD). Missense Mutation | Probably damaging |
| Patient 2 | Autosomal recessive (inherited) | Congenital absence of skin over the right lower limb. The healed wounds show hypo-pigmentation, milia and minimal atrophy. | Reduced staining of type VII collagen | p. Asp850Tyr and p. Leu2246Phe Missense mutation Two homozygous mutations in exon19 and 85. | Mutations (exon85) are damaging (score 1.0). The stability of the protein is decreased (SIFT) |
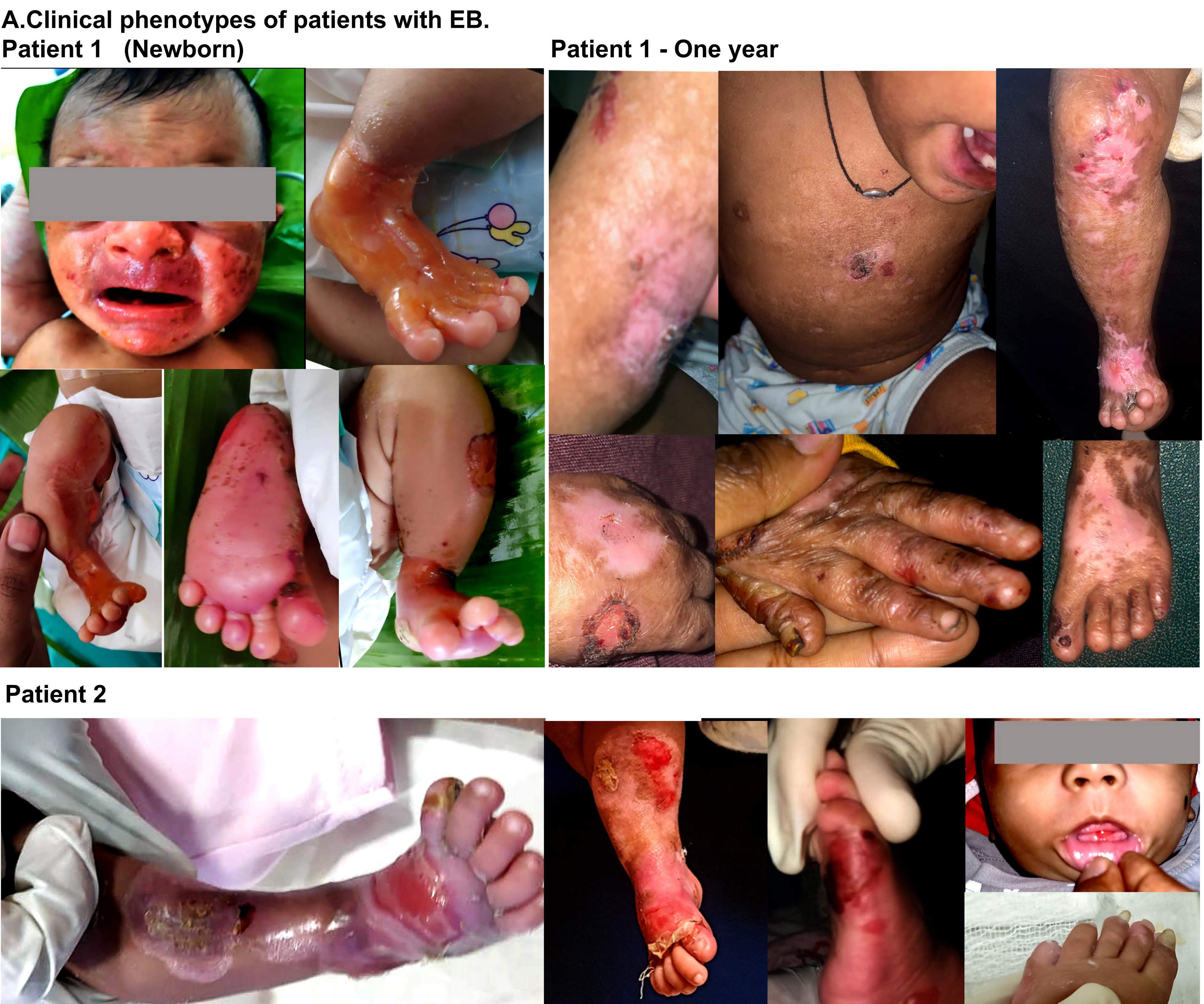
-
Clinical and molecular Investigations of Infants with ACC. A clinical feature showing blistering and erosion of the skin at birth (patient 1). The images of the wounds and deformity due to wound healing, scarring and fibrosis (patient 1-one year). The ACC in the new born with wound healing issues and recurrent blisters (patient 2).
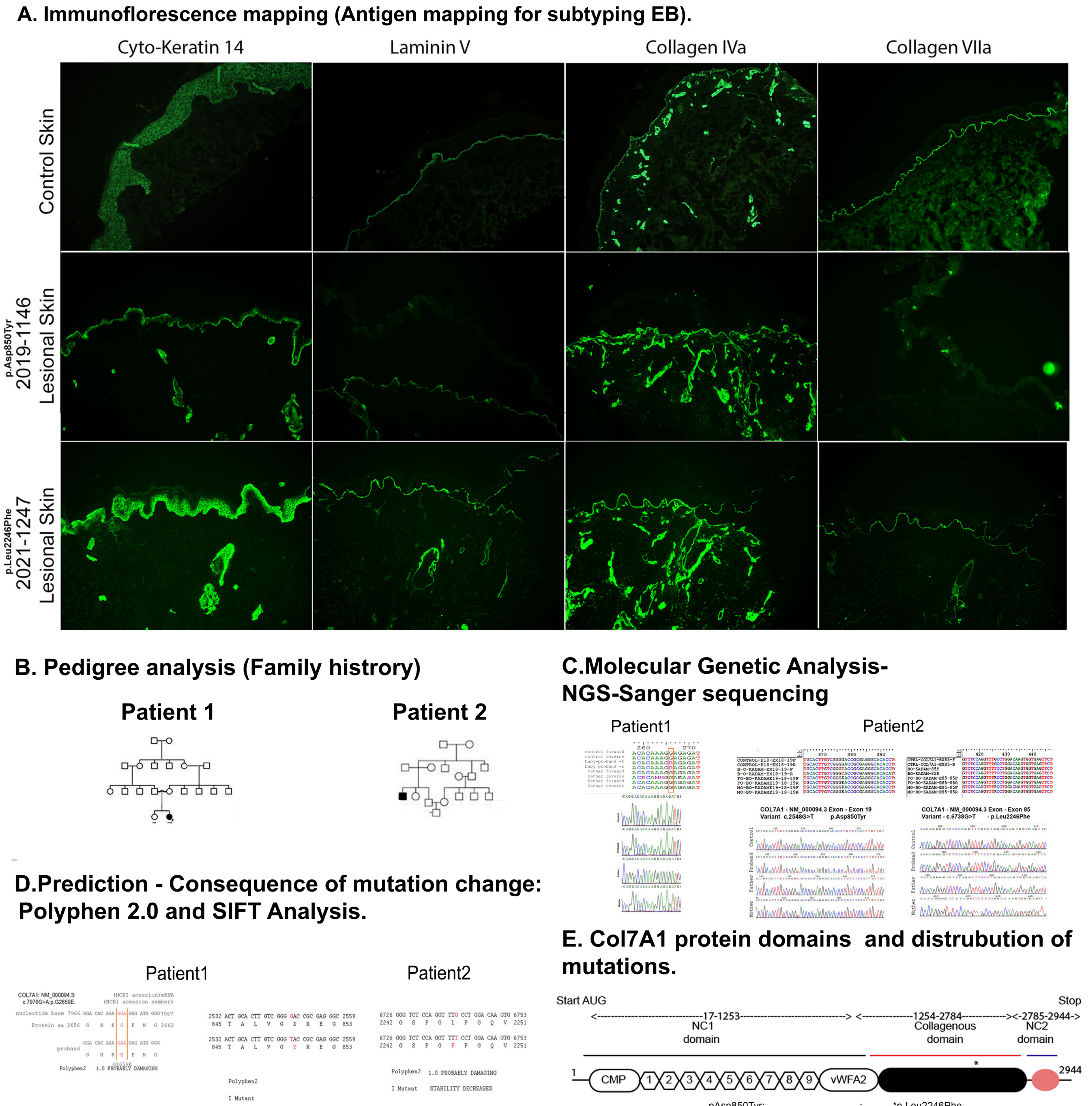
- Molecular Investigations for Dystrophic Epidermolysis Bullosa (DEB) patients. (A) Immunofluorescence mapping (IFM) of the skin from patient skin (magnification ×100). (B). Pedigree constructed based on family history. (C) Genetic analysis of the baby and parents for genetic loci G2659E, D850Y and L2246F. (D) The consequence of missense mutation and the effect on protein function is predicted using bioinformatics tools-SIFT and PolyPhen 2.0 E. The schematic diagram shows collagen 7A primary protein structure and its domains. The mutation G2659E and L2246F is in the triple helix domain of the protein.
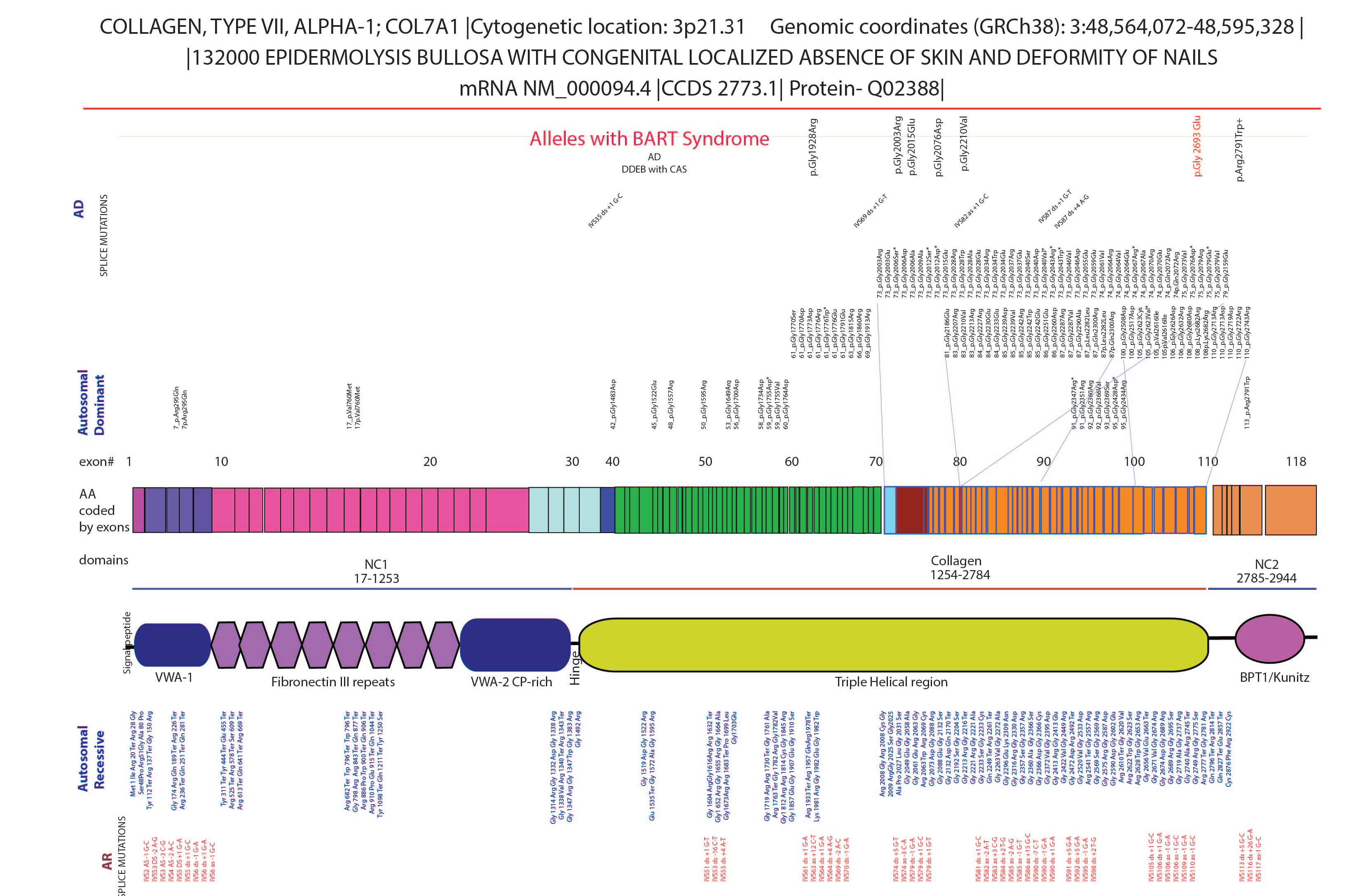
- The genetic locus of collagen VII allele. The mutations in the gene collagen 7a cause dystrophic epidermolysis bullosa. The large gene has 118 exons with both recessive and dominant dystrophic epidermolysis bullosa. The mutations in the triple helix are enriched for dystrophic epidermolysis bullosa. The alleles and reports describing aplasia cutis congenita are shown in the top row
Declaration of patient consent
The study is reviewed and approved by BMCRI Ethics Committee (BMCRI/PS/205).
Financial support and sponsorship
The Study is supported by grants from Department of Information Technology, Biotechnology and Science and Technology, Govt of Karnataka, and research grants from Rajiv Gandhi University of Health Sciences, Bangalore (17C014B).
Conflict of interest
There are no conflicts of interest.
References
- Biology of anchoring fibrils: Lessons from dystrophic epidermolysis bullosa. Matrix Biol. 1999;18:43-54.
- [CrossRef] [PubMed] [Google Scholar]
- Inversa dystrophic epidermolysis bullosa is caused by missense mutations at specific positions of the collagenic domain of collagen type VII. J Invest Dermatol. 2010;130:2508-11.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype-phenotype correlations on epidermolysis bullosa with congenital absence of skin: A comprehensive review. Clin Genet. 2021;99:29-41.
- [CrossRef] [PubMed] [Google Scholar]
- Injury- and inflammation-driven skin fibrosis: The paradigm of epidermolysis bullosa. Matrix Biol. 2018;68-69:547-60.
- [CrossRef] [PubMed] [Google Scholar]
- Dominant-negative effects of COL7A1 mutations can be rescued by controlled overexpression of normal collagen VII. J Biol Chem. 2009;284:30248-56.
- [CrossRef] [PubMed] [Google Scholar]




