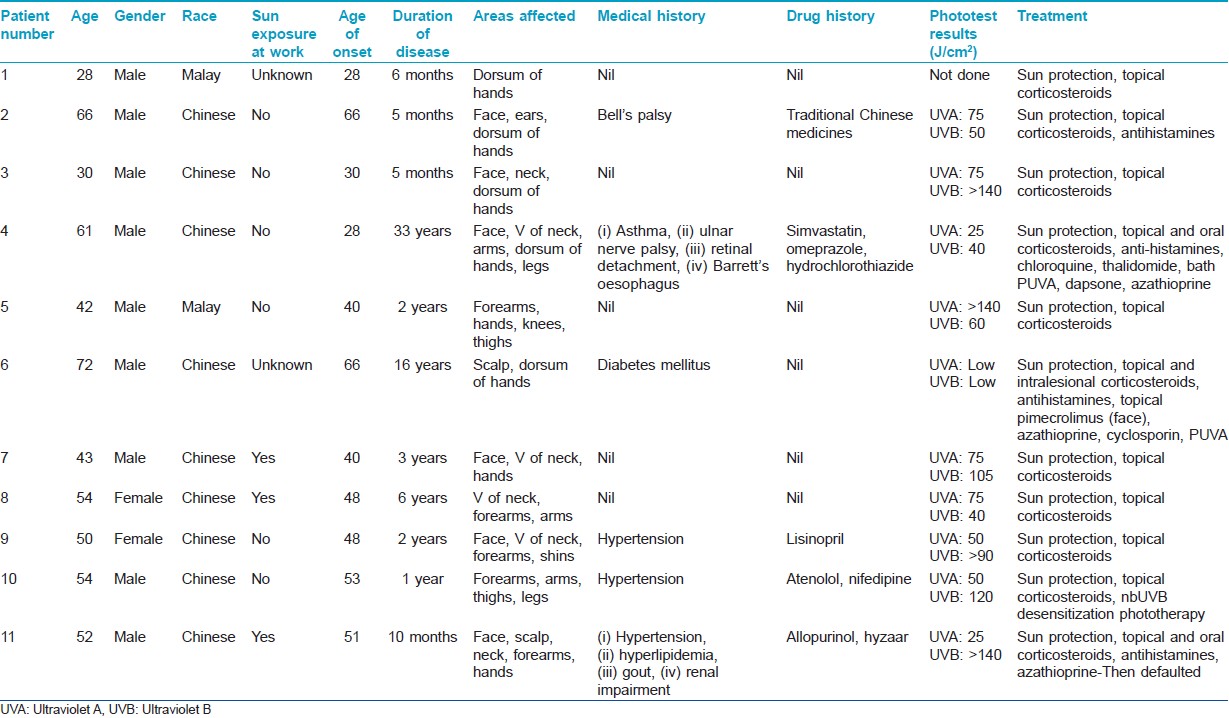
Translate this page into:
Clinical characteristics of adult-onset actinic prurigo in Asians: A case series
Correspondence Address:
Khor Jia Ker
Department of Dermatology, National Skin Centre, 1 Mandalay Road, 308205
Singapore
| How to cite this article: Ker KJ, Chong WS, Theng CS. Clinical characteristics of adult-onset actinic prurigo in Asians: A case series. Indian J Dermatol Venereol Leprol 2013;79:783-788 |
Abstract
Background: Actinic prurigo (AP) is a chronic, pruritic skin condition caused by an abnormal reaction to sunlight. Aims: The aim of this study is to determine the clinical characteristics of AP in patients attending the National Skin Centre, Singapore, from 1 st January 1999 to 30 th June 2008. Methods: Cases of AP diagnosed from 1 st January 1999 to 30 th June 2008 were retrieved from the center's electronic medical records and analyzed. Results: A total of 11 patients were diagnosed with AP. The mean age at diagnosis was 52 years. There were 9 (82%) Chinese and 2 (18%) Malay patients. Nine (82%) were male and 2 (18%) were female. The most commonly affected areas were the face, forearms, and hands (72%). Phototesting showed reduced minimal erythema dose (MED) to ultraviolet A (UVA) in 5 (46%) patients, both UVA and ultraviolet B (UVB) in 4 (36%) patients and UVB in 1 (9%) patient. Seven (64%) patients reported partial improvement after treatment with a combination of topical corticosteroids and sunscreens. Four (36%) patients received systemic therapy with partial response. Conclusion: Adult-onset AP is more common in the Asian population, with a male predominance. The face, forearms, and hands are the most commonly affected areas. The absence of mucosal involvement is also a distinguishing feature between the Asian and Caucasian population. Close to half of the patients have reduced MED to UVA on phototesting. The prognosis for AP is poor as it tends to run a chronic course with suboptimal response to treatment.Introduction
Actinic prurigo (AP) is a chronic, pruritic skin disease caused by an abnormal reaction to sunlight. AP can occur at any age, but usually begins before 10 years of age and often resolves in adolescence or early adult life. The eruption of AP is characterized by pruritic papules and nodules, which are frequently excoriated. Eczematization, lichenification, and crusting, with secondary infection are often present. [1] Cheilitis and conjunctivitis may also occasionally be present, particularly in native American patients. [2]
AP involves sun-exposed areas, such as the face (eyebrows, malar regions, nose, and lips), neck, V-areas of the chest, extensors of the arms and forearms as well as dorsum of the hands. It has been shown that these patients react to broad-spectrum radiation ultraviolet A (UVA) and ultraviolet B (UVB), but their minimal erythema dose (MED) to UVB and UVA are usually normal. [3]
Adult-onset AP, defined as the onset of AP after 21 years of age, has been reported and this variant is seen more commonly in Asian patients. Cheilitis and conjunctivitis are usually not associated with adult-onset AP. [4]
The aim of this study is to determine the clinical characteristics of patients with AP seen at the National Skin Center, a tertiary dermatology center in Singapore, from 1 st January 1999 to 30 th June 2008.
Methods
This study was approved by the local ethics review board. Cases of AP diagnosed from 1 st January 1999 to 30 th June 2008 were retrieved from electronic medical records in National Skin Center. Demographic data, including age, race, gender, occupation, onset and duration of disease, triggering factors, and clinical features were collated. We also reviewed all investigations performed, including blood tests, phototests, patch tests, and histopathology of skin biopsies. The responses to treatment in all patients were recorded.
Phototesting was carried out using the following light sources: A Kindermann slide projector equipped with a 150 W light bulb (Ochsenfurt, Germany) for visible light, a Supuvasun Mutzhas 3000 high-pressure metal-halide source (spectral output 350-450 nm, peak 370-385 nm) (Munich, Germany) for UVA, and a Dermaray M-DMR-100 bank of seven fluorescent bulbs (FL20S E-30/DMR 305 nm, emission spectrum 290-390 nm, peak 305 nm) (Eisai, Japan) for UVB. The patients′ buttocks were exposed to increasing doses of UV radiation. For UVA, radiation doses ranged in geometric progression from 25 J/cm 2 to 100 J/cm 2 (irradiance 24 mW/cm 2 at 21 cm distance) and for UVB, 30-200 mJ/cm 2 (irradiance 1 mW/cm 2 at 30 cm distance). The patients′ inner forearms were exposed to visible light emitted from the slide projector placed at a distance of 10 cm. The presence of wheals elicited by visible light, UVA or UVB were determined 20 min later and recorded. The MED responses for UVA and UVB were read at 24 h and the smallest dose to achieve "just perceptible erythema" is taken as the MED. For our local population and MED reading of less than 100 J/cm 2 to UVA and 100 mJ/cm 2 to UVB is considered abnormal. Monochromatic phototesting is not available at our center.
Results
A total of 11 patients were diagnosed to have AP from 1 st January 1999 to 30 th June 2008 [Table - 1]. Three of the patients had occupations with a significant sun exposure (patients 7, 8 and 11). There were 9 males (82%) and 2 females (18%). The mean age of presentation was 52 years (range: 28-72 years). Nine patients (82%) were Chinese and 2 (18%) were Malay. Two patients (18%) had a history of atopy and 2 (18%) had a family history of atopy.
Patients typically presented with excoriated papules and nodules, often with areas of lichenfication [Figure - 1] and [Figure - 2]. In our series, the most commonly affected areas were the face, forearms and hands as shown in 8 out of 11 patients (73%). This was followed by the legs in 7 patients (64%), neck in 6 (55%), arms in 5 (45%), V of the chest and trunk in 4 patients (36%). Only 3 patients (27%) had papules on the scalp, ears, and feet. None of the 11 patients had conjunctivitis or cheilitis.
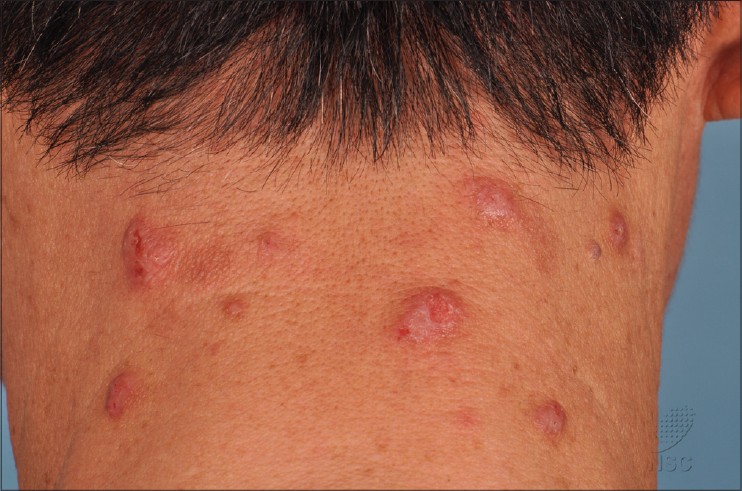 |
| Figure 1: Excoriated prurigo papules and nodules on the nape of the neck |
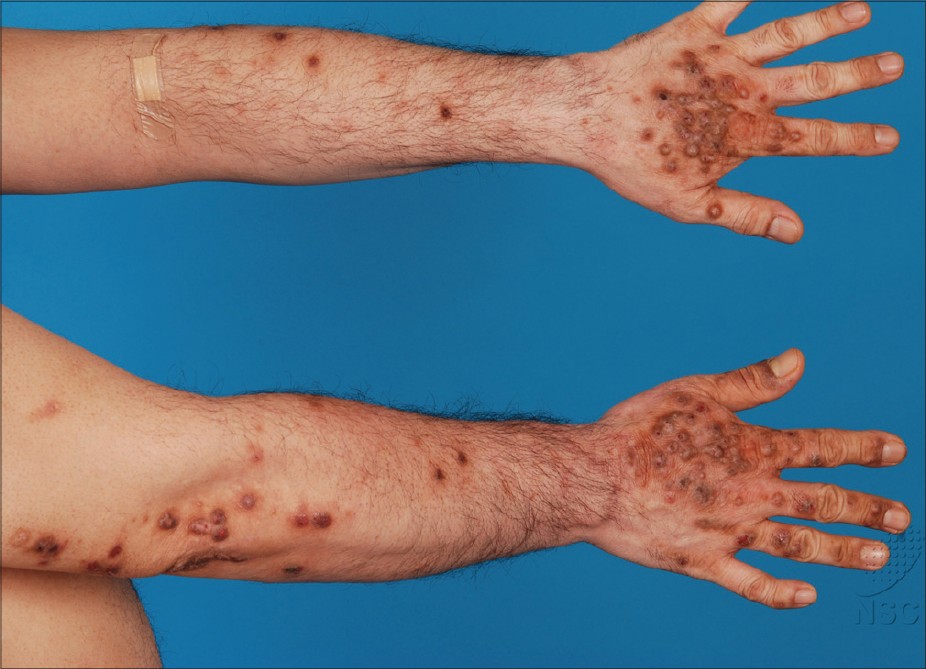 |
| Figure 2: Hyperpigmented excoriated nodules on areas of sun exposure-the forearms and dorsum of the hands |
The mean duration of disease at presentation was 6 years, with a range of 5 months to 33 years. The mean age at onset was 45 years (range: 28-66 years). Only 4 patients (36%) were aware that their lesions were photoaggravated. One patient reported a flare of his itchy rashes an hour after sun exposure with improvement in itch when he was in the shade. In the remaining three patients, their symptoms were aggravated by sun exposure and persisted even when sun exposure ceased. All patients had Fitzpatrick′s skin type IV.
Urinary and blood porphyrin screen were performed in 6 patients (55%) with negative results. Anti-nuclear antibodies were performed in 9 patients (82%). All had negative results except for 1 patient who had a low positive titer of 1:100. Tests for human immunodeficiency virus were not performed.
Phototesting was performed in the majority of patients (91%). The MED to UVA was decreased in 5 patients (46%), UVA and UVB in 4 (36%), and UVB in 1 patient (9%). None of the patients had a positive response to visible light. Photo patch test was performed in 1 patient with a negative result.
Four patients (36%) had skin biopsies taken from lesional skin. One biopsy showed mounds of parakeratosis, acanthosis, and slight basal layer hyperplasia. The dermis had increased superficial perivascular lymphocytic infiltrates. Three other biopsies showed non-specific changes.
All patients were treated with a combination of sunscreens and potent topical corticosteroids. Some patients had taken oral antihistamines with minimal or no improvement. Four patients (36%) received systemic therapy, including oral prednisolone, chloroquine, thalidomide, dapsone, azathioprine, cyclosporine, photochemotherapy with psoralens with ultraviolet A (PUVA) and UVA, and UVB phototherapy.
Treatment details of the 4 patients who received systemic therapy are elaborated below. Patient 4 was initially treated with chloroquine 200 mg 3 times a day for 2 months. However, this was stopped because of visual disturbances. He was then treated with oral prednisolone for more than a year. Thalidomide was added, starting at 100 mg daily and gradually reduced to 50 mg. As the lesions improved, prednisolone was tapered off. He relapsed 15 months later and thalidomide was restarted at 200 mg daily with good disease control. Thalidomide was continued for the next 3 years. He was subsequently treated with dapsone for a few months without improvement, and he was switched to azathioprine 50 mg daily, with gradual dose increment to 150 mg daily for 7.5 months. He responded well, but azathioprine was eventually stopped due to gastrointestinal side-effects and headache. Patient 6 was treated with azathioprine for a few months, but this was stopped due to side-effects. He was then switched to cyclosporine without improvement. The patient subsequently underwent PUVA therapy for 6 months with good results. Treatment was then changed to intralesional triamcinolone acetonide injection (10 mg/ml) and topical pimecrolimus on the face. Patient 10 was started on narrowband UVB phototherapy, but with minimal improvement. Patient 11 was treated with azathioprine 50 mg daily and prednisolone 40 mg daily, but he was irregular for follow-up visits.
These patients were followed-up for a mean duration of 48 months (range: 1 month to 20 years). Treatment could reduce the intensity of itch in most patients, achieving a partial improvement in symptoms. However, none of the patients achieved complete remission.
Discussion
AP is an immunologically mediated photodermatosis. [5],[6],[7] It is well-reported in the American Indian population, and affected individuals may have a genetic predisposition. Studies have reported a strong association between AP and the human leukocyte antigen (HLA)-DR4 allele, particularly subtype DRB1 FNx01 0407, which has been reported in a number of studies. [8],[9],[10],[11] In contrast, there have been no HLA associations with polymorphous light eruption, [9] which is a more common photodermatosis. The association between AP and HLA typing was not previously reported in the Asian population and as such, HLA typing was not performed in this series.
The relevance of an atopic history in AP is unknown. However, a personal or family history of atopy has been reported. [4] In our case series, 18% of patients had either a personal or family history of atopy, which was comparable to the general population.
Akaraphanth et al. [4] and Lestarini et al. [12] described adult-onset AP as being more common in the Asian population as opposed to the American population where AP usually starts in childhood. Adult-onset AP was also associated with a less favorable prognosis due to persistence of lesions. Our findings in this case series corroborate with previous studies in Asian populations, with a mean age at onset being 45 years and mean disease duration of 6 years, and a patient even had AP for up to 33 years.
There was a male predominance in our series with a ratio of 4.5:1. This is in contrast to findings by Ross et al. [13] in an Australian photobiology unit where there was a female preponderance.
Magaña et al. [14] reported that mucosal involvement was frequently associated with AP. In a cohort of 102 mestizo children, labial, and conjunctival mucosal lesions were found in 60% and 30% respectively. In another study by Ross et al., [13] cheilitis was found in 24% of Caucasian patients with AP. However in the Asian population, mucosal involvement was present in only 3% of cases in a study by Akaraphanth et al. [4] In our series, none of the patients had cheilitis or conjunctivitis, and this again differentiates the characteristics of AP between the Caucasian and Asian population.
Pruritus in AP is intense and chronic. This leads to chronic scratching, which in turn manifests as erythematous papules with crusts, excoriations, and lichenified plaques, [15] which may become secondarily infected. These typical manifestations were seen in our patients in a photo-distributed manner on the face, forearms, dorsum of hands and legs.
Phototesting in AP may show a reduction in MED to UVA, UVB or a combination of both. [16] In a series of 20 Australian patients, Crouch et al. [17] reported abnormal phototests in 60% of patients with AP. Forty percent of patients had a reduced MED to UVA and the remaining 20% had reduced MED to both UVA and UVB. However, in the United Kingdom, Grabczynska [18] found that 30% of patients had a decreased MED to UVB, 25% to both UVA and UVB, and only 5% of cases with a decreased MED to UVA. In our series, all AP patients who were tested had abnormal phototests. Results were similar to those of Crouch et al., [17] with close to half of the patients having reduced MED to UVA, 36% to both UVA and UVB and 9% to UVB alone.
Histology of lesional skin in AP is usually not helpful towards the diagnosis as findings are relatively non-specific. [13] However, Hojyo-Tomoka et al. [19] reported that AP has distinct histopathologic characteristics with hyperkeratosis, regular acanthosis, and dense infiltrates in the superficial dermis, composed mainly of lymphocytes. The biopsy from one of our patient showed similar findings to the above.
AP is a recalcitrant condition and treatment should be tailored to the individual patient. Essential points in management would include sun protection measures such as sun avoidance, protective clothing against ultraviolet radiation and broad-spectrum sunscreens. There is a paucity of treatment algorithms and of the numerous treatment modalities that have been utilized, thalidomide has proven its efficacy in most patients [20],[21],[22],[23] and appears to be superior when compared to other treatment options such as antimalarials, beta-carotene, antihistamines, azathioprine, cyclosporine, topical or systemic corticosteroids and phototherapy. However, adverse effects such as peripheral neuropathy and teratogenicity limit its use.
Our patients experienced partial improvement with topical and systemic treatments, but none achieved complete remission. Lane et al. [24] reported that 35% of his patients found no relief from any treatment and late-onset AP is less likely to improve with time. Only 17% of his patients improved within 5 years from the time they were first seen.
Conclusion
AP is an uncommon photodermatosis. The adult-onset variant of AP is more common in Asians compared to the Americans. A male predominance was seen in our series, in contrast to a female predominance world-wide. An atopic history was also not found to be significant. The absence of mucosal involvement in this series is another distinguishing feature between Asians and Caucasians. Close to half of the patients have a decreased MED to UVA alone on phototesting, consistent with previous reports. Treatment remains challenging and prognosis is guarded, with a chronic course of disease to be expected.
| 1. |
Rogers M, McCuaig C, Pruksachatkun C. Physical injury and environmental hazards. In: Schachner LA, Hansen RC, editors. Pediatric Dermatology. 3 rd ed. Edinburgh: Mosby; 2003. p. 1231-2.
[Google Scholar]
|
| 2. |
Hawk JL, Young A, Ferguson J. Abnormal cutaneous effects of UVR exposure: The photodermatoses. In: Burns T, Breathnach B, Cox N, Griffiths C, editors. Rook's Textbook of Dermatology. 8 th ed. Massachusetts: Blackwell Science Ltd.; 2010. p. 29.14.
th ed. Massachusetts: Blackwell Science Ltd.; 2010. p. 29.14.'>[Google Scholar]
|
| 3. |
Honigsmann H, Hojyo-Tomoka MT. Polymorphous light eruption, hydroa vacciniforme, and actinic prurigo. In: Lim HW, Hönigsmann H, Hawk JL, editors. Photodermatology. New York: Informa Healthcare; 2007. p. 160.
[Google Scholar]
|
| 4. |
Akaraphanth R, Sindhavananda J, Gritiyarangsan P. Adult-onset actinic prurigo in Thailand. Photodermatol Photoimmunol Photomed 2007;23:234-7.
[Google Scholar]
|
| 5. |
Moncada B, González-Amaro R, Baranda ML, Loredo C, Urbina R. Immunopathology of polymorphous light eruption. T lymphocytes in blood and skin. J Am Acad Dermatol 1984;10:970-3.
[Google Scholar]
|
| 6. |
Umaña A, Gómez A, Durán MM, Porras L. Lymphocyte subtypes and adhesion molecules in actinic prurigo: Observations with cyclosporin A. Int J Dermatol 2002;41:139-45.
[Google Scholar]
|
| 7. |
Estrada-G I, Garibay-Escobar A, Núñez-Vázquez A, Hojyo-Tomoka T, Vega-Memije E, Cortés-Franco R, et al. Evidence that thalidomide modifies the immune response of patients suffering from actinic prurigo. Int J Dermatol 2004;43:893-7.
[Google Scholar]
|
| 8. |
Batard ML, Bonnevalle A, Ségard M, Danzé PM, Thomas P. Caucasian actinic prurigo: 8 cases observed in France. Br J Dermatol 2001;144:194-6.
[Google Scholar]
|
| 9. |
Grabczynska SA, McGregor JM, Kondeatis E, Vaughan RW, Hawk JL. Actinic prurigo and polymorphic light eruption: Common pathogenesis and the importance of HLA-DR4/DRB1*0407. Br J Dermatol 1999;140:232-6.
[Google Scholar]
|
| 10. |
Suárez A, Valbuena MC, Rey M, de Porras Quintana L. Association of HLA subtype DRB10407 in Colombian patients with actinic prurigo. Photodermatol Photoimmunol Photomed 2006;22:55-8.
[Google Scholar]
|
| 11. |
Zuloaga-Salcedo S, Castillo-Vazquez M, Vega-Memije E, Arellano-Campos O, Rodríguez-Pérez JM, Pérez-Hernández N, et al. Class I and class II major histocompatibility complex genes in Mexican patients with actinic prurigo. Br J Dermatol 2007;156:1074-5.
[Google Scholar]
|
| 12. |
Lestarini D, Khoo LS, Goh CL. The clinical features and management of actinic prurigo: A retrospective study. Photodermatol Photoimmunol Photomed 1999;15:183-7.
[Google Scholar]
|
| 13. |
Ross G, Foley P, Baker C. Actinic prurigo. Photodermatol Photoimmunol Photomed 2008;24:272-5.
[Google Scholar]
|
| 14. |
Magaña M, Mendez Y, Rodriguez A, Mascott M. The conjunctivitis of solar (actinic) prurigo. Pediatr Dermatol 2000;17:432-5.
[Google Scholar]
|
| 15. |
Hojyo-Tomoka MT, Vega-Memije ME, Cortes-Franco R, Domínguez-Soto L. Diagnosis and treatment of actinic prurigo. Dermatol Ther 2003;16:40-4.
[Google Scholar]
|
| 16. |
Kuno Y, Sato K, Hasegawa K, Tsuji T. A case of actinic prurigo showing hypersensitivity of skin fibroblasts to ultraviolet A (UVA). Photodermatol Photoimmunol Photomed 2000;16:38-41.
[Google Scholar]
|
| 17. |
Crouch R, Foley P, Baker C. Actinic prurigo: A retrospective analysis of 21 cases referred to an Australian photobiology clinic. Australas J Dermatol 2002;43:128-32.
[Google Scholar]
|
| 18. |
Grabczynska SA, Hawk JL. What is actinic prurigo in Britain? Photodermatol Photoimmunol Photomed 1997;13:85-6.
[Google Scholar]
|
| 19. |
Hojyo-Tomoka MT, Dominguez-Soto L, Vargas-Ocampo F. Actinic prurigo: Clinical-pathological correlation. Int J Dermatol 1978;17:706-10.
[Google Scholar]
|
| 20. |
Yong-Gee SA, Muir JB. Long-term thalidomide for actinic prurigo. Australas J Dermatol 2001;42:281-3.
[Google Scholar]
|
| 21. |
Ng JC, Foley PA, Crouch RB, Baker CS. A case of severe actinic prurigo successfully treated with thalidomide. Australas J Dermatol 2001;42:192-5.
[Google Scholar]
|
| 22. |
Crouch RB, Foley PA, Ng JC, Baker CS. Thalidomide experience of a major Australian teaching hospital. Australas J Dermatol 2002;43:278-84.
[Google Scholar]
|
| 23. |
Holzer A, Wolf P, Legat FJ, Kerl H, Hofer A. Successful thalidomide therapy for actinic prurigo in a European woman. J Dtsch Dermatol Ges 2006;4:961-4.
[Google Scholar]
|
| 24. |
Lane PR, Hogan DJ, Martel MJ, Reeder B, Irvine J. Actinic prurigo: Clinical features and prognosis. J Am Acad Dermatol 1992;26:683-92.
[Google Scholar]
|
Fulltext Views
5,148
PDF downloads
1,332





![[Table - 1]](#tbl_ijdvl_2013_79_6_783_120726_t1.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2013_79_6_783_120726_f2.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_6_783_120726_f3.jpg){kind=link}