Translate this page into:
Clinical profile and mutation analysis of xeroderma pigmentosum in Indian patients
2 Department of Pediatrics, All India Institute of Medical Sciences, New Delhi, India
3 Department of Dermatology, BYL Nair Ch. Hospital and T. N. Medical College, Mumbai Central, India
4 Department of Dermatology, Grant Medical College and Sir JJ Hospital Group of Hospitals, Byculla, India
5 Department of Dermatology, Seth GS Medical College and KEM Hospital, Parel, Mumbai, India
6 Department of Pediatrics, BJ Wadia Hospital for Children, Parel, Mumbai, India
7 Department of Pediatrics, ESIS Hospital, Thane, Employees State Insurance Scheme Hospital, India
Correspondence Address:
Parag M Tamhankar
Scientist 'D', ICMR Genetic Research Centre, National Institute for Research in Reproductive Health, JM Street, Parel, Mumbai - 400 012
India
| How to cite this article: Tamhankar PM, Iyer SV, Ravindran S, Gupta N, Kabra M, Nayak C, Kura M, Sanghavi S, Joshi R, Chennuri VS, Khopkar U. Clinical profile and mutation analysis of xeroderma pigmentosum in Indian patients. Indian J Dermatol Venereol Leprol 2015;81:16-22 |
Abstract
Background: Xeroderma pigmentosum (XP) is an autosomal recessive genetic disorder characterized by cutaneous and ocular photosensitivity and an increased risk of developing cutaneous neoplasms. Progressive neurological abnormalities develop in a quarter of XP patients. Aim: To study the clinical profile and perform a mutation analysis in Indian patients with xeroderma pigmentosum. Methods: Ten families with 13 patients with XP were referred to our clinic over 2 years. The genes XPA, XPB and XPC were sequentially analyzed till a pathogenic mutation was identified. Results: Homozygous mutations in the XPA gene were seen in patients with moderate to severe mental retardation (6/10 families) but not in those without neurological features. Two unrelated families with a common family name and belonging to the same community from Maharashtra were found to have an identical mutation in the XPA gene, namely c.335_338delTTATinsCATAAGAAA (p.F112SfsX2). Testing of the XPC gene in two families with four affected children led to the identification of the novel mutations c.1243C>T or p.R415X and c.1677C>A or p.Y559X. In two families, mutations could not be identified in XPA, XPB and XPC genes. Limitation: The sample size is small. Conclusion: Indian patients who have neurological abnormalities associated with XP should be screened for mutations in the XPA gene.INTRODUCTION
Xeroderma pigmentosum (XP) is an autosomal recessive genetic disorder of DNA repair characterized by cutaneous and ocular photosensitivity and an increased risk of developing cutaneous neoplasms such as basal cell carcinoma, squamous cell carcinoma and melanoma. Progressive neurological abnormalities including deafness, spasticity and cognitive impairment may develop in about 25% of XP patients. Nine complementation groups have been described and mutations occur in any of the nine corresponding genes: XPA, ERCC3(XPB), XPC, ERCC2, DDB2, ERCC4, ERCC5, ERCC1 and POLH1. [1] Mutations arise most commonly in XPA (25%), XPC (25%), POLH1 (21%) or XPD (15%) genes. [2] In addition to the XP phenotype, mutations in genes causing XP are also associated with phenotypes such as trichothiodystrophy (TTD) (with or without XP), Cockayne syndrome (with or without XP) and cerebro-oculo-facial syndrome (COFS). [2] The frequency of XP in the United States and Europe is 1 in a million [3] while in Japan it has been reported as 1 in 22000. [4] The incidence in India is not known. A literature search identified several case reports from India but mutation analysis had not been performed in any of the cases. [5],[6],[7],[8],[9],[10] Till date, 37 unrelated families with patients having XP have been reported from India. Many families have been reported from South India especially Karnataka, where significant consanguinity is observed. [5],[6],[7],[8],[9],[10] In the present study, mutation analysis was performed for the XPA gene in eight families, the XPB gene in three of the eight families and the XPC gene in four families.
Clinical Report
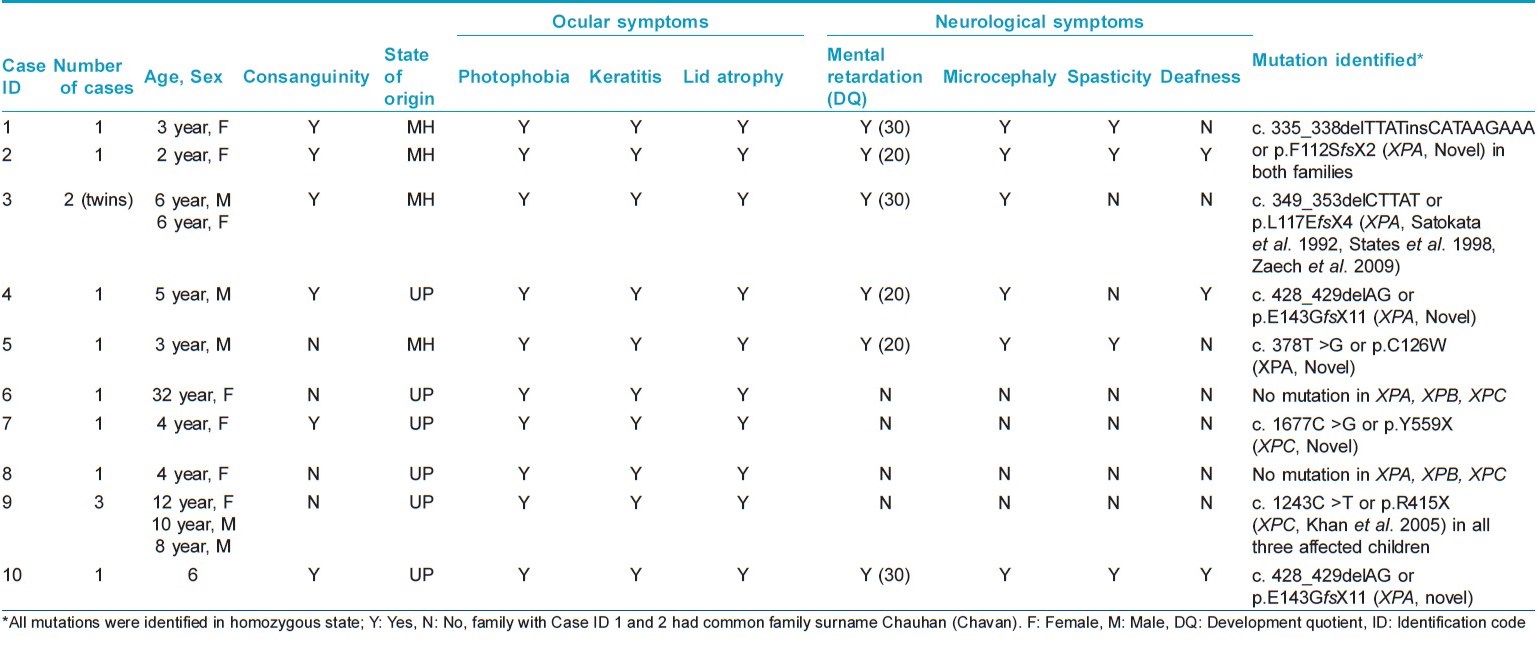
Thirteen patients from 10 unrelated families with XP were referred to the ICMR Genetic Research Center, Mumbai for molecular diagnosis and counseling during the period 2011-2014. Consanguinity was observed in 6 of the 10 families. Five families each belonged to Maharashtra and Uttar Pradesh. All XP patients showed typical clinical features of cutaneous photosensitivity such xerosis, poikiloderma and numerous freckle like hyperpigmented macules on sun-exposed skin. Cutaneous symptoms were noted by 6 months to 1 year of age. None of the patients had cutaneous neoplasms. Neurological features included mental retardation (6/13), ataxia (6/13), spasticity (3/13), microcephaly (6/13), and deafness (3/13). Computed tomography scan of the brain performed for one patient revealed cerebral atrophy with dilated lateral ventricles. Basal-evoked response audiometry done in two cases revealed sensorineural deafness. Photophobia was present in all patients while 6 patients each had keratitis and lid atrophy. All children had normal hair. Blistering and bleeding followed by pigmentation was observed in one 32 year old woman. Multiple hypopigmented macules along with lesions that worsened on sun exposure were also observed in this case. [Figure - 1] shows clinical, radiological, and pathological features of the patients. Clinical findings are summarized in [Table - 1].
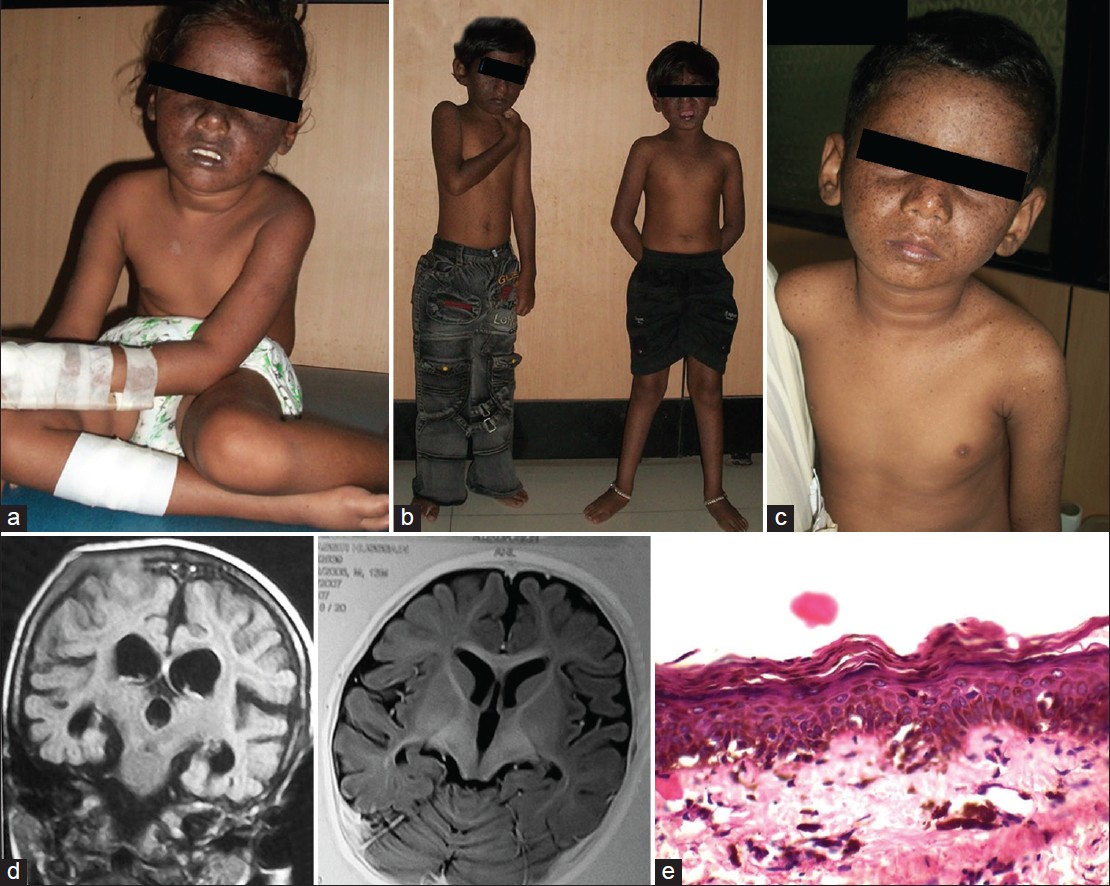 |
| Figure 1: (a-c) Patients with xeroderma pigmentosum, Case ID 1, Case ID 3 (twins) and Case ID 4, respectively. (d) MRI brain of Case ID 4 showing cerebral and cerebellar atrophy. (e) Skin biopsy: epidermis showing acanthosis with hyperpigmentation in the basal layer and presence of melanin incontinence (H and E stain) ×400 |
The patients were counseled to avoid sun exposure as far as possible and to use sunscreens especially during travel. Neurological problems were managed with physiotherapy. All families received genetic counseling and were explained the possibility of prenatal diagnosis in their future pregnancies.
METHODS
Informed consent for clinical photography and genetic testing was obtained from parents of affected children. The study was approved by the institutional ethics committee. Primer sequences were generated using Primer3 software version 0.4.0 available at http://frodo.wi.mit.edu/. [11] Mutation analysis by bidirectional Sanger sequencing of all exons and exon-intron boundaries was performed for the XPA gene in all index cases of nine families, of which three families were also tested for mutations in the XPB gene. The XPC gene was analyzed in the three index cases of the ninth family and for the index cases of the sixth, seventh and eighth families.
DNA amplification was performed for each fragment in a 10 μl final volume containing 100 ng genomic DNA, 1 mM dNTPs, 10 pmol of each primer, 1U Taq KAPA Hotstart polymerase and 1 μl 10X PCR buffer (KAPA Biosystems, MA, USA). Thirty-five cycles of amplification were used, each consisting of 1 min denaturation at 95°C, 45 s annealing at 60-65°C suitable for each primer set, 45 s extension at 72°C and final extension time of 10 min in a thermal cycler. The products were run on a 2% agarose gel along with the appropriate negative controls and a 100 bp ladder. Products that passed this quality check were purified using Exo-SAP-IT™ (USB Corporation, OH, USA) and then sequenced using BigDye Terminator v3.1 and capillary electrophoresis was performed using an automated sequencer ABI-3730XL (Applied Biosystems, CA, USA). A comparison with wild-type sequences was done using online BLAST software (blast.ncbi.nlm.nih.gov/Blast.cgi). The mutations were named according to GenBank accession Nos. NM_000380.3 and NM_001145769.1 for the XPA and XPC complementary DNA sequences and NP_000371.1 and NP_001139241.1 for the XPA and XPC protein sequences, respectively. Nomenclature and pathogenicity of the mutation were confirmed using Mutation T@ster software available at http://www.mutationtaster.org. [12] Nonsynonymous missense mutations were tested for their pathogenicity using SIFT (sift.jcvi.org) [13] and Polyphen2 (genetics.bwh.harvard.edu/pph 2/). [14]
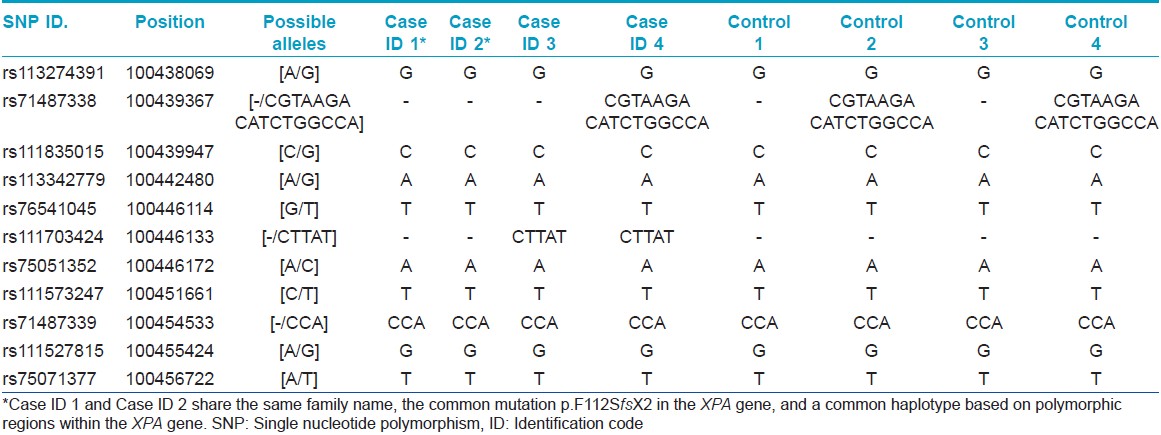
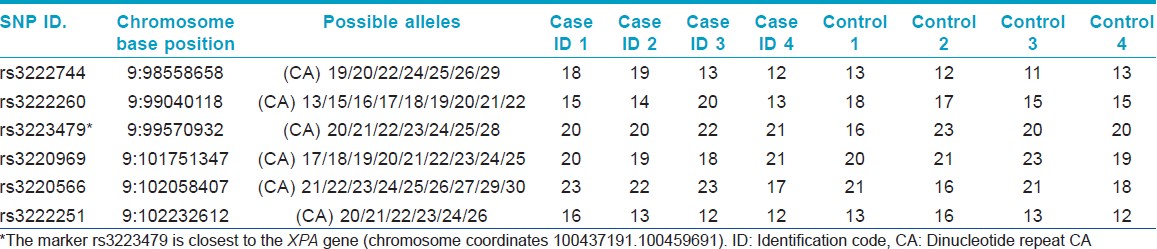
Haplotype analysis was performed for the mutation c. 335_338delTTATinsCATAAGAAA (p.F112SfsX2) by sequencing polymorphic regions of the XPA gene. CA repeat regions close to the XPA gene on chromosome number 9 were analyzed on an automated sequencer. The chromosomal loci of the SNPs and CA repeats are given in [Table - 2] and [Table - 3].
RESULTS
Four distinct mutations (three in exon 3 and one in exon 4) were found in the XPA gene in our patients. The novel mutation c. 378T > G or p.C126W in exon 3 was predicted to be pathogenic using bioinformatics analysis [SIFT score 0, PPH 2 1 (sensitivity 0, specificity 1) score, MT score 215]. Another novel homozygous mutation c. 1243C > T (p.R415X) was found in the XPC gene of one family. This patient also showed homozygosity for benign polymorphisms c. 1475G > A (p.R492H) and c. 1496C > T (p.A499V) in the XPC gene. A third novel mutation c. 1677C > G (p.Y559X) was found in the XPC gene in another family. Mutations in the XPA gene were found in patients with severe neurological abnormalities. Parents were confirmed as carriers for the same mutations. The mutation c. 335_338delTTATinsCATAAGAAA (p.F112SfsX2) was found in two unrelated families from Maharashtra (with the same family name Chauhan/Chavan) and founder effect was confirmed by haplotyping analysis as shown in [Table - 2] and [Table - 3]. Chromatograms of mutant and wild-type sequences are shown in [Figure - 2]. The mutation c. 428_429delAG (p.E143GfsX11) in exon 4 of XPA gene was also found in two unrelated families from Uttar Pradesh. Haplotype analysis could not be performed for this mutation.
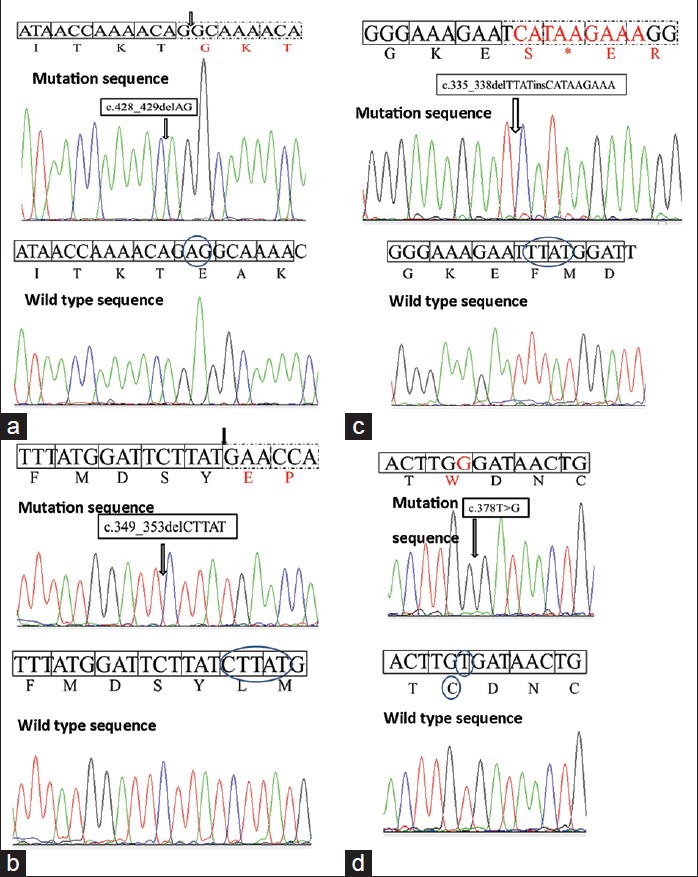 |
| Figure 2: (a-d) Shows the sequence chromatograms of mutant and wild-type sequences of XPA gene |
Prenatal diagnosis was offered to family 2 (case ID 2) and targeted mutation analysis was done on fetal DNA obtained after chorion villus biopsy. Maternal contamination was ruled out using short tandem repeat analysis using the commercial kit, Identifiler (Applied Biosystems). The fetus was identified to be a unaffected carrier for mutation. The diagnosis was confirmed to be accurate postnatally.
DISCUSSION
Differential diagnoses
XP was diagnosed in our cases based on the characteristic clinical findings. Trichothiodystrophy (TTD) was ruled out by the absence of ichthyosis, brittle hair and facial features of protruding ears and micrognathia. In trichothiodystrophy, hair microscopy under polarized light shows a characteristic dark and light transverse banding pattern known as "tiger tail banding." [15] Xeroderma pigmentosum/Cockayne syndrome (XP/CS) may be differentiated from XP by its typical physical appearance of "cachectic dwarfism" with thinning of the skin and hair, sunken eyes, and a stooped standing posture. Cataracts, pigmentary retinopathy and ataxia may be present in these patients. [16] Cerebrooculofacial syndrome (COFS) also needs to be distinguished from XP; this condition presents with failure of growth, microcephaly with intracranial calcifications, microcornea, cataracts, optic atrophy and congenital joint contractures, while some of the characteristic features of XP (xerosis, poikiloderma, atrophy and telangiectasia) are absent. [17]
Patients with XPA mutations usually have mild to severe neurological abnormalities. Mutations in the ERCC3 gene (XPB group) may account for the XP/CS phenotype, TTD and XP with mild neurologic abnormalities. Patients with mutations in the XPC gene usually do not have neurological abnormalities. Patients with mutations in ERCC2 (XPD group) may have either XP (with or without neurological involvement), XP/CS, XP/TTD, TTD or COFS phenotypes. Mutations in the DDB2 gene (XPE group) lead to XP with no neurological abnormality. Mutations in ERCC4 (XP F group) causes XP with no neurologic abnormalities or severe late-onset neurologic abnormalities. Mutations in the ERCC5 gene (XP G group) are seen in XP with no neurologic abnormalities or severe neurologic abnormalities or may lead to XP/CS phenotype. Mutations in ERCC1 may be found in patients with COFS. The XP variant phenotype does not have neurological involvement and is caused by mutations in the POLH gene. [2]
Genetics
The Human Gene Mutation Database (www.hgmd.cf.ac.uk) [18] lists 32 unique XPA and 55 unique XPC mutations whereas the XP mutation database ( http://xpmutations.org ) [1] lists 128 mutant alleles of the XPA and 54 mutant alleles of the XPC gene. Most mutations reported are point mutations or splice site mutations; [19] however, these were not identified in our study. Three of the four mutations were frameshift. These mutations (namely c. 335_338delTTATinsCATAAGAAA (p.F112SfsX2) and c. 349_353delCTTAT (p.L117EfsX4) in exon 3 and c. 428_429delAG (p.E143GfsX11) in exon 4) resulted in a premature stop codon, thereby resulting in mRNA instability due to nonsense-mediated decay. This may explain the severe phenotype in our patients. The fourth mutation, namely c. 378T > G (p.C126W) in exon 3 was a missense mutation.
The mutation delCTTAT has been identified earlier in a compound heterozygosity with c. 288delT in the XPA gene in an XP-affected uncle-nephew pair. Both of them showed XP with severe neurological deterioration. [20] The mutation was also found in a homozygous state in a Caucasian patient having XP with severe neurological deterioration. [21]
All patients with confirmed XPA gene mutations showed signs of severe neurological abnormalities whereas in two patients with no neurological manifestations, homozygous mutations c. 1243C > T (p.R415X) and c. 1677C > G (p.Y559X), were identified in the XPC gene. Both mutations were premature chain termination mutations. The p.R415X mutation was considered as causative in the ninth family as the peptide chain would be prematurely terminated before the other mutation sites. This mutation was previously observed in a 10-year-old male of French origin without neurological involvement. [22]
Founder mutations in XP
The rare mutation c. 335_338delTTATinsCATAAGAAA (p.F112SfsX2) found in two unrelated families with the same family name Chauhan (Chavan), points toward a founder effect as confirmed by haplotyping analysis. Chauhan (Chavan) is a Marathi surname of Rajput origin. [23] The Marathi speaking Chauhan (Chavan) clan belongs to the 96 clan Marathas who were the fighters/rulers in medieval era. [24] They are descendants of the great Prithviraj Chauhan of Ajmer, Rajasthan. [25] Further patients belonging to this group need to be studied to confirm these observations and to know the carrier rate.
There are several published observations of population specific founder mutations for XP. The founder mutation IVS3-1G > C in the XPA gene accounts for approximately 90% of alleles in Japan. The carrier rate for this mutation in Japan is as high as 1 in 100. [4] Soufir et al. (2010) identified the c. 1643_1644delTG mutation in the XPC gene as the major mutation in XP patients from North Africa (Morocco, Tunisia and Algeria) (74% of overall XP and 87% in XPC type). [26] Masaki et al. (2008) identified four founder mutations accounting for 87% of XP variant type in Japan. [27]
CONCLUSIONS
Indian patients of xeroderma pigmentosum presenting with neurological symptoms should be screened for mutations in the XPA gene. Rapid molecular diagnosis would aid definitive diagnosis, genetic counseling and prenatal diagnosis.
ACKNOWLEDGEMENT
The authors acknowledge the families for their participation and acknowledge Dr. Siv Tang from the DNA Diagnostics Lab, Boston Children′s hospital, USA for performing mutation analysis of XPC gene in Case ID 9.
| 1. |
Cleaver JE, Thompson LH, Richardson AS, States JC. A summary of mutations in the UV-sensitive disorders: Xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat 1999;14:9-22.
[Google Scholar]
|
| 2. |
Kraemer KH, DiGiovanna JJ. Xeroderma Pigmentosum. 2003. Gene Reviews™. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1397/[Last accessed on 2013 Aug 05].
[Google Scholar]
|
| 3. |
Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A, et al. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 2008;7:744-50.
[Google Scholar]
|
| 4. |
Hirai Y, Kodama Y, Moriwaki S, Noda A, Cullings HM, Macphee DG, et al. Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res 2006;601:171-8.
[Google Scholar]
|
| 5. |
Kulkarni ML, Rani RS. Xeroderma pigmentosum. Indian Pediatr 1996;33:394-8.
[Google Scholar]
|
| 6. |
Mane DR, Kale AD, Hallikerimath S, Angadi P, Kotrashetti V. Trichilemmal carcinoma associated with xeroderma pigmentosa: Report of a rare case. J Oral Sci 2010;52:505-7.
[Google Scholar]
|
| 7. |
Raju MS, Suma GN, Ravi Prakash SM, Goel S. Xeroderma Pigmentosum: Variable Expressions among Three Siblings. J Indian Acad Oral Med Radiol 2010;22:109-12.
[Google Scholar]
|
| 8. |
Grampurohit VU, Dinesh US, Rao R. Multiple cutaneous malignancies in a patient of xeroderma pigmentosum. J Cancer Res Ther 2011;7:205-7.
[Google Scholar]
|
| 9. |
Sharma S, Deshmukh AD, Bal MM, Chaukar DA, Dcruz AK. Angiosarcoma of the scalp associated with Xeroderma pigmentosum. Indian J Med Paediatr Oncol 2012;33:126-9.
[Google Scholar]
|
| 10. |
Bandyopadhyay R, Nag D, Bandyopadhyay S, Sinha SK. Atypical fibroxanthoma: An unusual skin neoplasm in xeroderma pigmentosum. Indian J Dermatol 2012;57:384-6.
[Google Scholar]
|
| 11. |
Rozen S, Skaletsky HJ. Primer3 on the WWW for general users and for biologistprogrammers. In: Krawetz S, Misener S, editors. Bioinformatics Methods and Protocols: Mehtods in Molecular Biology. Totowa, NJ: Humana Press; 2000. p. 365-86.
[Google Scholar]
|
| 12. |
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575-6.
[Google Scholar]
|
| 13. |
Ng PC, Henikoff SS. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812-4.
[Google Scholar]
|
| 14. |
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248-9.
[Google Scholar]
|
| 15. |
Liang C, Kraemer KH, Morris A, Schiffmann R, Price VH, Menefee E, et al. Characterization of tiger tail banding and hair shaft abnormalities in trichothiodystrophy. J Am Acad Dermatol 2005;52:224-32.
[Google Scholar]
|
| 16. |
Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology 2000;55:1442-9.
[Google Scholar]
|
| 17. |
Graham JM, Anyane-Yeboa K, Raams A, Appeldoorn E, Kleijer WJ, Garritsen VH, et al. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am J Hum Genet 2001;69:291-300.
[Google Scholar]
|
| 18. |
Krawczak M, Cooper D. The human gene mutation database. Trends Genet 1997;13:121-2.
[Google Scholar]
|
| 19. |
States JC, McDuffie ER, Myrand SP, McDowell M, Cleaver JE. Distribution of Mutations in the Human Xeroderma Pigmentosum Group A Gene and Their Relationships to the Functional Regions of the DNA Damage Recognition Protein. Hum Mut 1998;12:103-13.
[Google Scholar]
|
| 20. |
Christen-Zaech S, Imoto K, Khan SG, Oh KS, Tamura D, Digiovanna JJ, et al. Unexpected Occurrence of Xeroderma Pigmentosum in an Uncle and Nephew. Arch Dermatol 2009;145:1285-91.
[Google Scholar]
|
| 21. |
Satokata I, Tanaka K, Okada Y. Molecular basis of group a xeroderma pigmentosum: A missense mutation and two deletions located in a zinc finger consensus sequence of the XPAC gene. Hum Genet 1992;88:603-7.
[Google Scholar]
|
| 22. |
Khan SG, Oh K, Shahlavi T, Ueda T, Busch DB, Inui H, et al. Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis 2005;27:84-94.
[Google Scholar]
|
| 23. |
Gordon S. The marathas 1600-1818. Cambridge: Cambridge University Press; 1993. p. 46. ISBN 978-0-521-26883-7. Retrieved 16 July 2011.
[Google Scholar]
|
| 24. |
Aiyappan LK, Ratnam B. Society in India. Chennai, Social Sciences Association; 1956. p. 41.
[Google Scholar]
|
| 25. |
Gupta RK, Bakshi SR, editors. Studies in Indian History: Rajasthan through the ages: The Heritage of Rajputs. New Delhi, Sarup and Sons. 2008. p. 95.
[Google Scholar]
|
| 26. |
Soufir N, Ged C, Bourillon A, Austerlitz F, Chemin C, Stary A, et al. A prevalent mutation with founder effect in xeroderma pigmentosum group C from North Africa. J Invest Dermatol 2010;130:1537-42.
[Google Scholar]
|
| 27. |
Masaki T, Ono R, Tanioka M, Funasaka Y, Nagano T, Moriwaki S, et al. Four types of possible founder mutations are responsible for 87% of Japanese patients with Xeroderma pigmentosum variant type. J Dermatol Sci 2008;52:144-8.
[Google Scholar]
|
Fulltext Views
5,406
PDF downloads
3,235





![[Figure - 1]](#fig_ijdvl_2015_81_1_16_148559_f3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2015_81_1_16_148559_t1.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2015_81_1_16_148559_t2.jpg){kind=link}
![[Table - 3]](#tbl_ijdvl_2015_81_1_16_148559_t5.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_1_16_148559_f4.jpg){kind=link}