
Translate this page into:
Compound heterozygosity in type-II oculocutaneous albinism
Corresponding author: Dr. Shibhani Sudheer Hegde, Department of Dermatology, Venereology and Leprosy, Bharati Vidyapeeth Medical College and Hospital, Pune, Maharashtra, India. shibhani.s.hegde@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Doshi S, Sawarthia S, Hegde SS, Sardesai VR. Compound heterozygosity in type-II oculocutaneous albinism. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_1121_2024
Dear Editor,
Oculocutaneous Albinism (OCA) is a group of genetically heterogeneous autosomal recessive (AR) disorders associated with mutations in genes controlling the biosynthesis of melanin. Classification of OCA has morphed from phenotypic to genotypic stratification. Other than OCA1A, which presents with a complete absence of melanin in the skin, hair, and eyes; other subtypes can present with wide phenotypic heterogeneity making differentiation challenging.1 Hence, genetic analysis plays a crucial role in accurate diagnosis, prognosis, and genetic counselling.
A six-month-old girl, first child of a third-degree consanguineous marriage, presented with light-coloured skin and hair since birth. This presentation was not stark when compared to her parents. Parents also reported nystagmus since birth and occasional photosensitivity but no history of seizures, recurrent infections, breathlessness, bleeding tendencies, photophobia, or hearing difficulties. Birth and developmental history were uneventful. The phenotype of the father was normal; the mother exhibited blue-grey irides. A detailed pedigree [Figure 1] revealed light-coloured skin and eyes in one of the paternal cousins, in whom there was a subsequent darkening of skin colour.
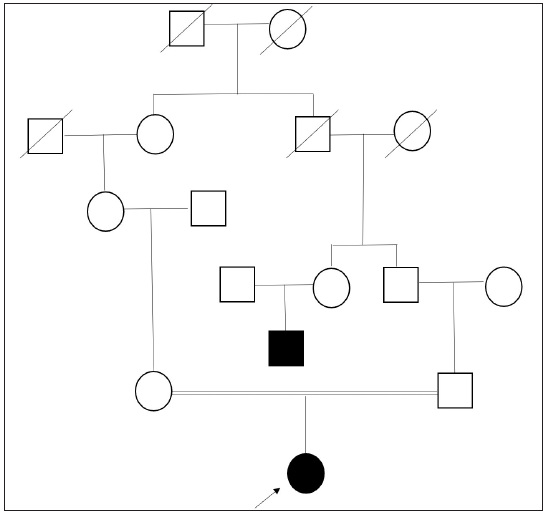
- Pedigree chart exhibiting third-degree consanguinity with affected proband and her paternal uncle. (Square: male; Round: female; Strike-through: deceased; Blacked out: affected.)
Physical examination revealed diffuse hypopigmented skin [Figure 2a] that made naevus simplex over the occiput and glabella more prominent [Figure 2b]. Scalp hair and eyelashes were dark brown, eyebrows were light brown, and blue-grey irides. Hair shaft examination revealed a uniform reduction in pigmentation without melanin clumps or structural defects [Figure 2c], and ophthalmologic evaluation demonstrated bilateral nystagmus and hypopigmented fundus.
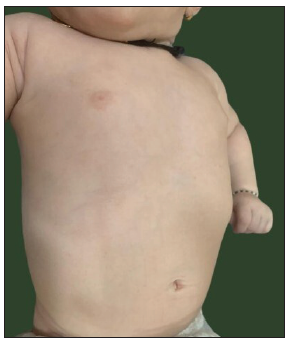
- Diffuse pigment dilution showing light skin over chest and abdomen.
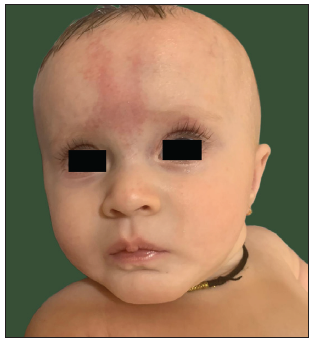
- Generalised pigment dilution of face making glabellar naevus simplex more pronounced.
![Hair shaft examination showing diffuse reduction of pigment along the shaft with no structural abnormalities or melanin clumps [Light microscopy, 40x].](/content/126/2025/0/1/img/IJDVL_1121_2024-g4.png)
- Hair shaft examination showing diffuse reduction of pigment along the shaft with no structural abnormalities or melanin clumps [Light microscopy, 40x].
After obtaining a written informed consent, clinical exome sequencing using next-generation sequencing (NGS) was performed that showed compound heterozygous mutations in the OCA2 gene. A novel 5’-splice-site variant in intron 10 (chr15:g.27990575C>T) and previously reported missense mutation in exon 19 (chr15:g.2792686G>C) were noted.
Splice-site mutation (c.1116+1G>A) affected the donor splice site downstream of exon 10 and was not reported in 1000 genome, gnomAD (v3.1, v2.1) and Topmed databases with the in-silico prediction of ‘damaging’ by MutationTaster2.
A missense mutation (c.2020C>G, p.Leu674Val) resulted in the amino acid substitution of valine with leucine at codon 674 at the citrate transporter domain of the OCA2 protein, which was previously reported in homozygous and compound heterozygous states.1
No other pathogenic variants were noted in other genes governing pigmentation. No other contiguous micro-deletions leading to Angelman syndrome or Prader-Willi syndrome were noted either. These variants could not be confirmed with Sanger sequencing due to financial constraints. Nevertheless, family was counselled regarding inheritance patterns and need for Sanger sequencing should the next pregnancy be planned. Strict photo-protection and regular ophthalmological evaluation were also emphasised.
Table 1 highlights the various subtypes of albinism and other related disorders.1-5
|
Oculocutaneous albinism (OCA) AR inheritance |
OCA1 |
Mutation in TYR gene Broadly divided into OCA1A and OCA1B; includes previously described temperature-sensitive OCA, minimal pigment OCA, and yellow OCA. OCA1A: complete inactivity of enzyme resulting in absent melanin in eye, skin, and hair OCA1B: reduced enzyme activity resulting in variable morphology |
| OCA2 |
Mutation in the OCA2 gene (formerly called the P gene) Includes brown OCA |
|
| OCA3 |
Mutation in TYRP1 gene Includes red or rufous OCA |
|
| OCA4 |
Mutation in SLC45A2/MATP gene Phenotype similar to OCA2 |
|
| OCA5 | Mutation in an unknown gene on locus 4q24 | |
| OCA6 | Mutation in SLC24A5 gene | |
| OCA7 | Mutation in C10orf11/LRMDA gene | |
| OCA8 | Mutation in DCT/TYRP2 gene | |
|
Ocular albinism (OA) X-linked recessive inheritance |
Non-syndromic disorder of ocular pigment not affecting skin and hair Divided into OA1 and OA2 |
|
|
Proposed syndromic types of albinism AR inheritance with mutation of molecules involved in generation of lysosome-related organelles (LROs) |
Hermansky-Pudlak syndrome |
OCA with bleeding diathesis, and specific organ involvement due to ceroid-lipofuscin deposition Includes 11 subtypes associated with mutations in 11 different genes |
| Silvery gray hair syndromes |
Chediak–Higashi syndrome: hypopigmentation of skin, hair, and eyes, silvery sheen of hair, recurrent infections, progressive neurologic abnormalities, mild coagulation defects, and a high risk of developing hemophagocytic lymphohistiocytosis Griscelli syndrome: hypopigmentation, with or without neurological abnormalities/immunodeficiency depending on the specific genetic defect |
|
|
Others: Cross-McKusick-Breen syndrome: oculocerebral syndrome with hypopigmentation and abnormalities of the central nervous system Cutaneous albinism, delayed myelination, and development, organomegaly due to mutations in the CLCN7 gene MAPBP-interacting protein deficiency syndromes due to mutations in the LAMTOR2 gene resulting in oculocutaneous albinism, short stature, and immunodeficiency |
||
AR: Autosomal recessive, TYR: Tyrosinase, OCA2: Oculocutaneous albinism, TYRP1: Tyrosinase related protein 1, SLC45A2: Solute carrier family 45 member 2, MATP: Membrane-Associated transporter protein, SLC24A5: Solute carrier family 24 member 5, C10orf11/LRMDA: Leucine rich melanocyte differentiation associated, DCT/TYRP2: Dopachrome tautomerase, CLCN7: Chloride voltage-gated channel 7, LAMTOR2: Late endosomal/lysosomal adaptor, MAPK and MTOR activator 2, MAPBP: Mitogen-activated protein-binding protein
OCA2 is caused by a mutation in the OCA2 gene (located on 15q12-q13.1chromosome containing 838 amino acids over 30 exons), which encodes for the P-protein. P-protein contains 12 transmembrane domains, which are crucial for normal biogenesis of melanosomes by maintaining acidic pH and for processing and transporting TYR and TYRP1.6 Maternal and paternal micro-deletions on chromosome 15q can result in Angelman syndrome and Prader-Willi syndrome due to their proximity to the OCA2 gene.2
Patients with OCA2 present with minimal to near normal cutaneous pigmentation at birth as melanocytes retain some ability to synthesise melanin (tyrosinase-positive). Irides colour varies from blue to brown. Overall pigmentation may turn darker with age, and despite subsequent improvement, they may develop consequences secondary to chronic sun damage. Additionally, visual problems, although less severe than OCA1, can be highly debilitating and are an essential manifestation in early diagnosis of OCA2.6 Therefore, regular use of sunscreen, protective clothing, and periodic dermatological and ophthalmological evaluation is imperative.
Clinical heterogeneity in OCA and diversity within mutation of the OCA2 gene (homozygous or compound heterozygous), to possible overlap with Angelman syndrome or Prader-Willi syndrome establishes the pre-eminent roles of mutational analysis in albinism.
Compound heterozygosity entails the presence of two different mutated alleles at a particular gene locus and differs from homozygous mutations that necessitate acquiring two identical mutated alleles of a gene at similar loci, thus resulting in a more severe phenotype.7
All patient characteristics (including skin, hair, and eye colour) and mutations have been summarised in the supplementary table from a comprehensive PubMed search for ‘oculocutaneous albinism’, ‘compound heterozygous,’ and ‘OCA2 gene’.
Despite OCA2 being a milder phenotype of OCA, ophthalmological morbidity can be significant if ignored. Mutational analysis ensures a discussion regarding inheritance patterns, recurrence risk, reproductive options and helps clear doubts of worrying parents. With gene therapies making headway in genodermatoses, identification of the affected gene ensures specific treatment options in the future.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Molecular basis of albinism in India: Evaluation of seven potential candidate genes and some new findings. Gene. 2012;511:470-4.
- [CrossRef] [PubMed] [Google Scholar]
- Prader-Willi and angelman syndromes: Mechanisms and management. Appl Clin Genet. 2023;16:41-52.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Genetics of non-syndromic and syndromic oculocutaneous albinism in human and mouse. Pigment Cell Melanoma Res.. 2021;34(4):786-99.
- [CrossRef] [PubMed] [Google Scholar]
- Syndromic albinism: a review of genetics and phenotypes. Dermatol Online J.. 2003;9(5):5.
- [CrossRef] [PubMed] [Google Scholar]
- Lysosomal Storage and Albinism Due to Effects of a De Novo CLCN7 Variant on Lysosomal Acidification. Am J Hum Genet.. 2019;104(6):1127-38.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Genetic analysis of albinism caused by compound heterozygous mutations of the OCA2 gene in a Chinese family. Hereditas. 2024;161:8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Mutational analysis of the TYR and OCA2 genes in four chinese families with oculocutaneous albinism. PLoS One. 2015;10:e0125651.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




