Translate this page into:
Congenital cellular plexiform schwannoma mimicking a vascular lesion: Potential pitfalls in clinical and histopathological assessment
2 Department of Dermatology and Venereology, All India Institute of Medical Sciences, New Delhi, India
Correspondence Address:
Asit R Mridha
Department of Pathology, All India Institute of Medical Sciences, Ansari Nagar, New Delhi - 110 029
India
| How to cite this article: Nambirajan A, Mridha AR, Kumar P, Ray R. Congenital cellular plexiform schwannoma mimicking a vascular lesion: Potential pitfalls in clinical and histopathological assessment. Indian J Dermatol Venereol Leprol 2016;82:79-81 |
Sir,
Malignant peripheral nerve sheath tumors are rare in childhood and are usually associated with neurofibromatosis type 1. They present as a painful mass or sudden enlargement in a preexisting neurofibroma and show high mitosis, necrosis, vascular invasion, recurrence, metastasis and poor survival. Cellular plexiform schwannoma is a benign cellular nerve sheath tumor seen in children and unrelated to neurofibromatosis type 1 but frequently associated with increased mitosis and rapid growth raising the possibility of misdiagnosis as a malignant tumor.[1]
A 7-month-old male infant presented with a gradually progressing swelling on the left shoulder since birth. His family members had no stigmata of neurofibromatosis type 1. Physical examination revealed a 10 cm × 4 cm erythematous, telangiectatic, non-tender, firm, bosselated, nodulo-plaque on the left arm and shoulder [Figure - 1]. A clinical diagnosis of tufted angioma or a Kaposiform hemangioendothelioma was considered. Biopsy revealed variably sized, circumscribed nodules surrounded by a loose fibro-connective tissue in the deep dermis and composed of closely packed fascicles of monomorphic spindle cells with nuclear hyperchromasia and indistinct nucleoli [Figure - 2]a and [Figure - 2]b. Verocay bodies and Antoni B areas were not present. Mitotic index ranged up to 10 per 10 high power fields. No necrosis or thick-walled hyalinized blood vessel was seen. The tumor cells were diffusely immunopositive with S100 [Figure - 2]c and negative for smooth muscle actin, HMB-45, CD99, CD34, CD31, pan-cytokeratin and Melan-A. Ki-67 labeling index was approximately 35–40% [Figure - 2]d.
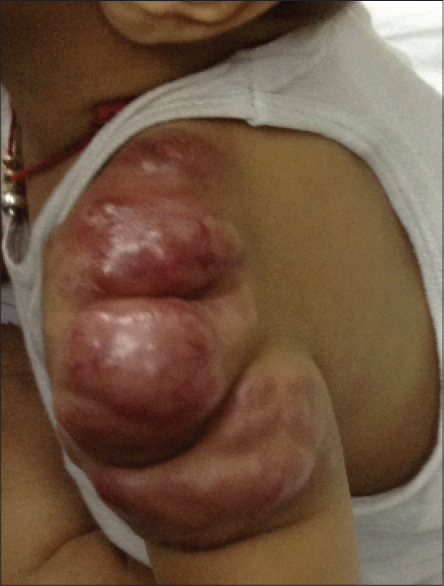 |
| Figure 1: Lobulated swelling with erythema and telangiectasia of the overlying skin |
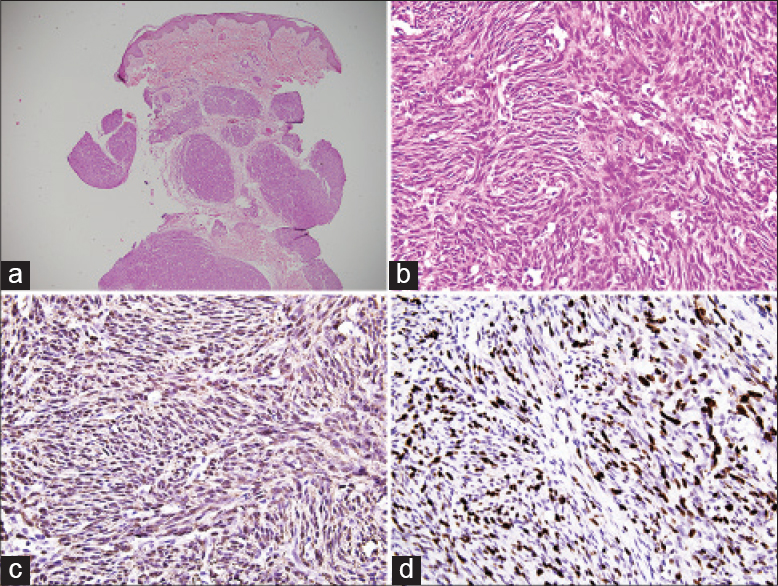 |
| Figure 2: (a) Plexiform and multinodular architecture without any capsule (H and E; ×40). (b) The nodules are composed of hypercellular spindle cells in fascicles with mitotic figures (H and E; ×400). (c) Spindle cells are diffusely immunopositive with S100 (×400). (d) Ki-67 labeling index is approximately 35–40% (×400) |
Schwannomas are usually solitary, sporadic tumours that affect all ages with a peak incidence in the fourth to sixth decades. Plexiform schwannoma involves multiple nerve fascicles or a nerve plexus with a plexiform, multinodular growth pattern and accounts for only 5% of all schwannomas. Nearly 90% are present on the head, neck, trunk, and upper extremities involving the small nerves of the skin and subcutaneous tissue. About 5% of cases are associated with neurofibromatosis type 2 and schwannomatosis.[2] Childhood plexiform schwannomas are rare; they are mostly seen from birth and occur on the lower extremity. As in our patient, a previously reported case was also mistaken for hemangioma.[3] Microscopically, schwannomas have Antoni A areas containing compact, palisading spindle cells with Verocay bodies alternating with Antoni B tissue exhibiting loosely arranged cells, lipid-laden histiocytes and thick-walled, hyalinized blood vessels. Childhood plexiform schwannomas contain mostly Antoni A tissue with increased mitotic activity and are devoid of Antoni B areas.[3],[4]
Meis-Kindblom and Enzinger in 1994 reported nine cases of “plexiform malignant nerve sheath tumor of infancy and childhood,” which showed brisk mitosis and high proliferation indices.[4] Eight of them were unrelated to neurofibromatosis type 1 and had local recurrences but a better survival rate compared to malignant peripheral nerve sheath tumors.[4] Woodruff et al. studied six histologically similar pediatric tumors and found that none of the patients suffered from metastatic disease or death.[1] Subsequently, these tumors were interpreted as plexiform cellular schwannomas.[1] Brisk mitosis and high proliferation indices in cellular plexiform schwannoma often lead to misdiagnosis of malignant peripheral nerve sheath tumor. Cellular plexiform schwannoma differs from schwannoma and non-plexiform cellular schwannoma by the lack of a well-formed capsule and degenerative changes.[1] Plexiform neurofibroma, which often is associated with neurofibromatosis type 1 and has a potential for malignant transformation, is composed of differentiated Schwann cells, fibroblasts, perineural-like cells, mast cells and myxoid stroma. Malignant peripheral nerve sheath tumor exhibits cellular atypia, mitosis, necrosis, vascular invasion and at least focal loss of S100 immunopositivity, whereas plexiform neurofibroma shows S100 immunopositivity only in 40–50% of the cells. In our case, the tumor was unencapsulated and showed no pleomorphism, necrosis or vascular invasion. However, it was diffusely immunopositive with S100. Other differential diagnoses include fibrosarcoma and dermatofibrosarcoma protruberans. Infantile fibrosarcoma is composed of minimally pleomorphic, mitotically active spindle cells. However, it shows no multinodularity and S100 positivity. Dermatofibrosarcoma protruberans has a characteristic storiform pattern, CD34 immunopositivity and is negative with S100.
Plexiform schwannoma shows near-diploid karyotypes with simple numerical changes often involving chromosomes 22, 7 and the sex chromosomes. Trisomy 17 has also been described.[5] Ultrastructurally, the tumor shows a continuous pericellular basal lamina.[4]
Local excision is the treatment of choice and is associated with a high local recurrence rate due to incomplete excision.[2] Except for death in a single patient with locally invasive orbital schwannoma, death or metastatic disease is virtually never seen.
Childhood cellular plexiform nerve sheath tumor often shows hypercellularity, alarmingly high mitotic activity and proliferative indices. However, the absence of cellular pleomorphism, necrosis and vascular invasion, especially when there is diffuse S100 positivity, should alert the pathologist to the lesion being benign.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Woodruff JM, Scheithauer BW, Kurtkaya-Yapicier O, Raffel C, Amr SS, LaQuaglia MP, et al. Congenital and childhood plexiform (multinodular) cellular schwannoma: A troublesome mimic of malignant peripheral nerve sheath tumor. Am J Surg Pathol 2003;27:1321-9.
[Google Scholar]
|
| 2. |
Agaram NP, Prakash S, Antonescu CR. Deep-seated plexiform schwannoma: A pathologic study of 16 cases and comparative analysis with the superficial variety. Am J Surg Pathol 2005;29:1042-8.
[Google Scholar]
|
| 3. |
Lo S, How P, Moss AL. Plexiform schwannoma mimicking haemangioma: Pitfalls in clinical diagnosis and histological interpretation. Br J Dermatol 2007;157:838-9.
[Google Scholar]
|
| 4. |
Meis-Kindblom JM, Enzinger FM. Plexiform malignant peripheral nerve sheath tumor of infancy and childhood. Am J Surg Pathol 1994;18:479-85.
[Google Scholar]
|
| 5. |
Tassano E, Sementa AR, Tavella E, Garaventa A, Panarello C, Morerio C. Trisomy 17 in congenital plexiform (multinodular) cellular schwannoma. Cancer Genet Cytogenet 2010;203:313-5.
[Google Scholar]
|
Fulltext Views
4,108
PDF downloads
2,195





![[Figure - 1]](#fig_ijdvl_2016_82_1_79_171012_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2016_82_1_79_171012_f2.jpg){kind=link}