Translate this page into:
Congenital erythropoietic porphyria in three siblings
Correspondence Address:
Arfan Ul Bari
Combined Military Hospital, Muzaffarabad, AJK
Pakistan
| How to cite this article: Bari A. Congenital erythropoietic porphyria in three siblings. Indian J Dermatol Venereol Leprol 2007;73:340-342 |
Abstract
Congenital erythropoietic porphyria is a rare autosomal recessive disorder that usually presents with marked skin photosensitivity, hypertrichosis, blistering, scarring, milia formation and dyspigmentation of the photo-exposed areas. Three adult siblings (two sisters and one brother) are presented here with variable degree of skin manifestations. During early childhood, all the siblings started showing signs of photosensitivity with darkening of urine color followed by skin blistering over the face and hands. The oldest showed severe sclerodermiform mutilation and the youngest exhibited an initial involvement with hypertrichosis. None of them had any history of convulsions, acute abdominal pain or joint pain. Woods lamp examination and laboratory investigations confirmed the diagnosis.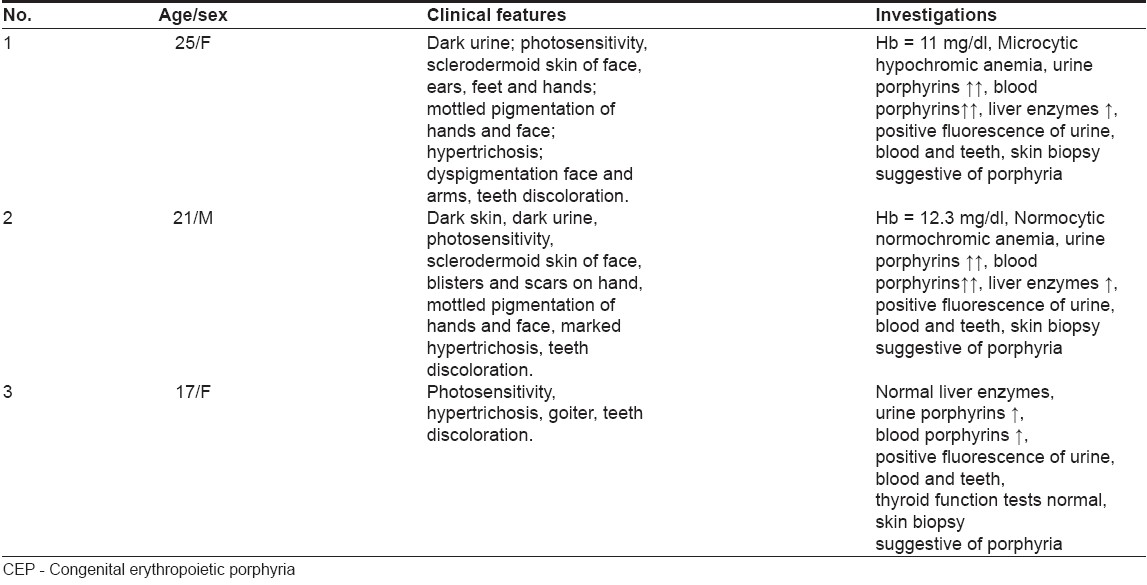
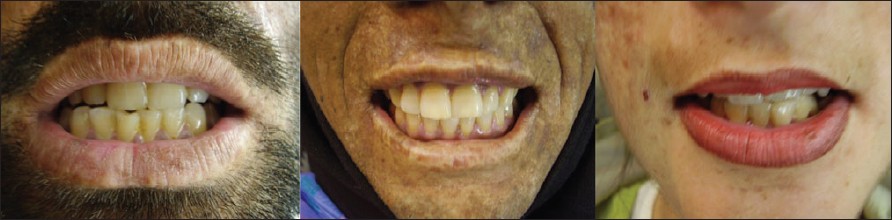 |
| Figure 3: Yellowish brown staining of teeth in 3 siblings |
|
| Figure 3: Yellowish brown staining of teeth in 3 siblings |
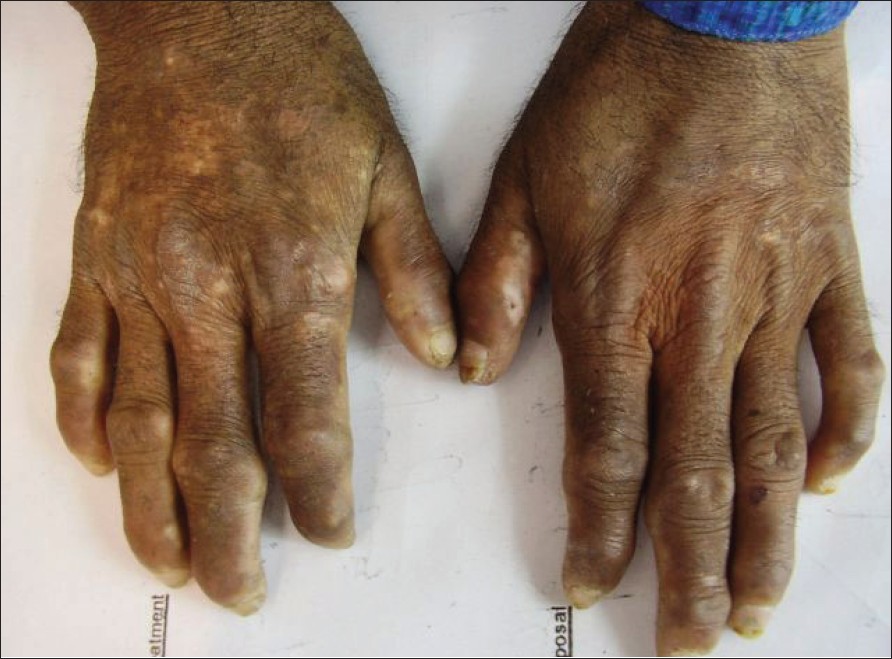 |
| Figure 2: Short stubby hands with hypertrichosis and scarring |
|
| Figure 2: Short stubby hands with hypertrichosis and scarring |
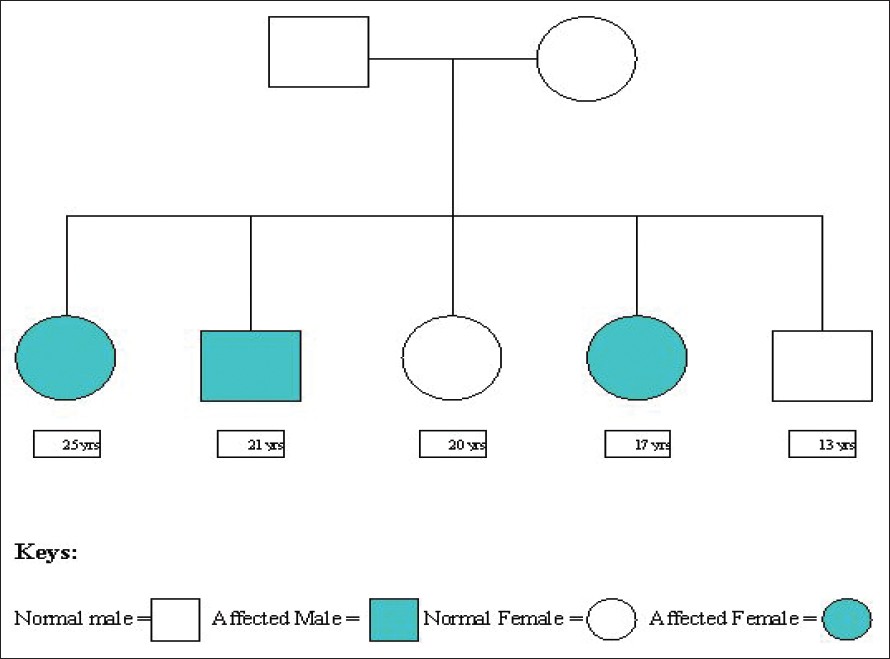 |
| Figure 1: Pedigree chart of the family |
|
| Figure 1: Pedigree chart of the family |
Introduction
Congenital erythropoietic porphyria (CEP), also known as Günther′s disease, is a rare inborn error of porphyrin-heme synthesis inherited as an autosomal recessive trait. It typically begins in infants or young children. The presentation of CEP at birth in a patient with history of a difficult perinatal course and concomitant jaundice usually indicates a severe disease. [1],[2] Despite the limited treatments that are currently available, the prognosis is not invariably poor. This rare disorder is reported here in three siblings of a family with non affected parents. The basic aim of presenting the cases was to highlight the diversity of clinical features and to report this uncommon occurrence simultaneously in three siblings.
Case Reports
Three patients (two sisters and one brother) were among the five siblings. They (first, second and fourth in the birth order) were the products of a consanguineous marriage. Both the parents and the other two siblings were healthy having no stigmata of the disease [Figure - 1].
Case 1
A 25-year-old single female had a history of dark urine and sensitivity to sunlight from her infancy. At the age of 7 years, a change in the color of teeth and appearance of blisters and vesicles on sun-exposed areas was observed. Gradually, the areas that developed repeated blisters became sclerotic. Clinical findings [Figure - 2],[Figure - 3] and laboratory investigations are shown in [Table - 1]. Skin biopsy from the hand showed epidermal atrophy with prominent dermal fibrosis and the absence of cutaneous adnexal structures. PAS staining showed the presence of an amorphous substance in the vessel walls.
Case 2
Case 2 was the younger brother of the abovementioned patient and had almost similar features with more pronounced hypertrichosis, vesicles, erosions, dyspigmentation and sclerosis of the face, ears and nose. Clinical findings [Figure - 2] and laboratory data are shown in [Table - 1].
Case 3
Case 3 was the youngest of the three affected siblings, a 17-year-old female who started having mild symptoms in her late childhood. It was three years ago that she started noticing significant hypertrichosis and photosensitivity over her face along with occasional blistering and ulceration over her hands. She incidentally was also found to have euthyroid goiter. Clinical findings and investigative data are shown in [Table - 1].
Discussion
CEP is a rare subtype of porphyria that causes the skin to be overtly sensitive to sunlight. Underlying defect is mutations of two alleles for the gene encoding the enzyme uroporphyrinogen III synthase that leads to the accumulation of the porphyrins of type-I isomer. The C73R mutation is the most frequent, in which cysteine is substituted by arginine. The accumulated isomer I porphyrinogens are spontaneously oxidized to their corresponding porphyrins, which are biologically useless but cause cutaneous photosensitivity characterized by blisters, erosions and scarring of light-exposed skin. [3],[4] These porphyrins are released from the maturing erythrocytes into the plasma and are excreted in urine, thereby producing a port-wine color. The interaction of excess porphyrins in the skin and light radiation causes photooxidative damage, manifesting as mechanical fragility and blistering. [1],[2],[3] Symptoms that appear in adulthood tend to be milder than symptoms that begin early in life.
Photosensitivity occurs early in the course of the disease. Increased fragility and erosions can contribute to mutilation, especially on the face and hands. Hypertrichosis of the face and extremities is common. The teeth have a reddish color [Figure - 3] and fluoresce under Wood′s lamp due to porphyrin deposition in the dentine and enamel. Ocular manifestations include blepharitis, cicatricial ectropion and conjunctivitis. Subsequent bilateral corneal scarring may occur, with eventual blindness. Porphyrins are also deposited in the bone resulting in loss of bone; subsequent contractures and deformities occur in adults with CEP. [1],[2],[3] In more serious cases, the hemolytic anemia of CEP can cause hypersplenism. [4] Bright pink fluorescence of urine, teeth and bones under Wood′s lamp illumination can help in diagnosis. The brilliant fluorescence of nuclei in erythrocyte precursor cells is specific for EP. [5]
Skin biopsy shows subepidermal blister with superficial perivascular lymphocytic infiltrate. Blood vessels have thickened, hyalinized periodic acid-Schiff (PAS) positive walls. Another interesting feature is festooning of papillary dermal tips into the blister cavity. Direct immunofluorescence tests reveal linear C3 and IgG staining around the superficial vessels and along the dermal-epidermal junction. [5],[6]
For management of such patients, absolute avoidance of sun exposure is crucial and sunscreens containing zinc oxide or titanium dioxide are preferable. Oral photoprotectants (beta-carotene) may prevent tissue damage due to light exposure, possibly by forming an internal light screen. Splenectomy may be indicated for intractable hemolytic anemia. Transfusion of erythrocytes, intravenous hematin, oral activated charcoal and cholestyramine, bone marrow transplant and gene therapy are other options. [6],[7],[8]
The patients described (three siblings) were among the five children of a consanguineous marriage. In two of them, the disease manifested early during infancy. They had short stubby fingers with mutilation of the hands along with fresh blistering and background sclerodermoid skin changes on face and hands. One of them had relatively adult onset disease with less-pronounced cutaneous features. There are two main differential diagnoses: erythropoietic protoporphyria (EPP) and hepato-erythropoietic porphyria (HEP). [9] EPP was ruled out on the basis of positive fluorescence of urine under Wood′s lamp. Similarly, HEP was excluded as both uroporphyrins and coproporphyrins were elevated and protoporphyrins were normal. However, the best confirmatory test to differentiate these would be high pressure liquid chromatography (HPLC), which was not available in our setup. All the patients had significant cutaneous involvement, but fortunately none had any serious internal problems such as hemolytic anemia, splenomegaly and bone or eye involvement. The cutaneous involvement can be managed in such cases by maximum abstinence from exposure to the sun as it is also important to prevent cutaneous neoplasms, which are one of the complications of CEP. [10] Thus far, around 200 cases have been reported worldwide and most of these were described as a single case. The disease is occasionally reported in two siblings; however, to the best of our knowledge, this is the first report of the occurrence of CEP in three siblings. [11]
| 1. |
Murphy GM. The cutaneous porphyrias: A review. The British Photodermatology Group. Br J Dermatol 1999;140:573-81.
[Google Scholar]
|
| 2. |
Fritsch C, Bolsen K, Ruzicka T, Goerz G. Congenital erythropoietic porphyria. J Am Acad Dermatol 1997;36:594-610.
[Google Scholar]
|
| 3. |
Desnick RJ, Astrin KH. Congenital erythropoietic porphyria. advances in pathogenesis and treatment. Br J Haematol 2002;117:779-95.
[Google Scholar]
|
| 4. |
Bishop DF, Johansson A, Phelps R, Shady AA, Ramirez MC, Yasuda M, et al . Uroporphyrinogen III synthase knock-in mice have the human congenital erythropoietic porphyria phenotype, including the characteristic light-induced cutaneous lesions. Am J Hum Genet 2006;78:645-58.
[Google Scholar]
|
| 5. |
Zaider E, Bickers DR. Clinical laboratory methods for diagnosis of the porphyrias. Clin Dermatol 1998;16:277-93.
[Google Scholar]
|
| 6. |
Murphy GM. Diagnosis and management of the erythropoietic porphyrias. Dermatol Ther 2003;16:57-64.
[Google Scholar]
|
| 7. |
Mathews-Roth MM. Treatment of the cutaneous porphyrias. Clin Dermatol 1998;16:295-8.
[Google Scholar]
|
| 8. |
Dupuis-Girod S, Akkari V, Ged C, Galambrun C, Kebaili K, Deybach JC, et al . Successful match-unrelated donor bone marrow transplantation for congenital erythropoietic porphyria (Gunther disease). Eur J Pediatr 2005;164:104-7.
[Google Scholar]
|
| 9. |
Massod QA, Hassan I, Khan D, Sameem F, Quadri MI, Hussain ST, et al . Congenital erythropoietic porphyria with hemolytic anemia. Indian J Dermatol 2005;50:155-7.
[Google Scholar]
|
| 10. |
Pandhi D, Suman M, Khurana N, Reddy BS. Congenital erythropoietic porphyria complicated by squamous cell carcinoma. Pediatr Dermatol 2003;20:498-501.
[Google Scholar]
|
| 11. |
Jacobo A, Almeida HL Jr, Jorge VM. Congenital erythropoietic porphyria in two siblings. Dermatol Online J 2005;11:15.
[Google Scholar]
|
Fulltext Views
2,810
PDF downloads
1,489





![[Figure - 1]](#fig_ijdvl_2007_73_5_340_35737_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2007_73_5_340_35737_2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2007_73_5_340_35737_3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2007_73_5_340_35737_4.jpg){kind=link}