Translate this page into:
Congenital generalized lipodystrophy of Berardinelli-Seip type: A rare case
2 Department of Endocrinology, All India Institute of Medical Sciences, New Delhi, India
Correspondence Address:
Sujay Khandpur
Department of Dermatology and Venereology, All India Institute of Medical Sciences, New Delhi
India
| How to cite this article: Khandpur S, Kumar A, Khadgawat R. Congenital generalized lipodystrophy of Berardinelli-Seip type: A rare case. Indian J Dermatol Venereol Leprol 2011;77:402 |
Abstract
Congenital generalized lipodystrophy of Berardinelli-Seip type is a rare autosomal recessive disorder characterized by nearly complete absence of adipose tissue and a consequent generalized muscular appearance, which is recognized easily at birth. The condition is associated with various dermatological and systemic manifestations. We report a case of this form of lipodystrophy. The patient had several cutaneous manifestations, including severe acanthosis nigricans, generalized hyperpigmentation, curly scalp hair, prominent subcutaneous veins, and enlarged clitoris. She also had associated celiac disease.Introduction
Congenital generalized lipodystrophy, a rare autosomal recessive disorder originally described by Berardinelli and Seip, [1] has been reported in approximately 250 patients of various ethnic origins. The estimated worldwide prevalence is 1 in 10 million population. [2] The characteristic clinical feature is nearly complete absence of adipose tissue and, consequently, a generalized muscular appearance, which is recognized easily at birth. This is associated with various dermatological, musculoskeletal, visceral, metabolic, reproductive, and cardiac manifestations. The metabolic abnormalities may prove fatal and so necessitate optimal therapeutic and preventive measures. [3] Berardinelli-Seip congenital lipodystrophy (BSCL) must be differentiated from premature aging syndromes as there are several overlapping features, including lipodystrophy. We report a case of BSCL in view of its rarity. The patient had several cutaneous manifestations.
Case Report
A 7-year-old girl was referred from the Department of Endocrinology with complaints of progressive loss of subcutaneous fat and poor weight gain since 2-3 years despite having a voracious appetite. She had history of recurrent episodes of diarrhea since the age of 1 year. Family history was not significant.
On examination, she had generalized loss of subcutaneous fat, with prominent subcutaneous veins; there was severe acanthosis nigricans involving all flexures, generalized hyperpigmentation, curly scalp hair, a protuberant abdomen with hepatosplenomegaly, and cliteromegaly [Figure - 1]. She also had prominent pectoral and thigh muscles and enlarged joints of fingers and toes. Her height was 114 cm (below the 50 th percentile of the Agarwal growth chart) and the body mass index (BMI) was 12.5 kg/m 2 . She had no mental retardation. Cardiovascular evaluation was normal. Bone age was 6-7 years (by Grulich and Pyle method).
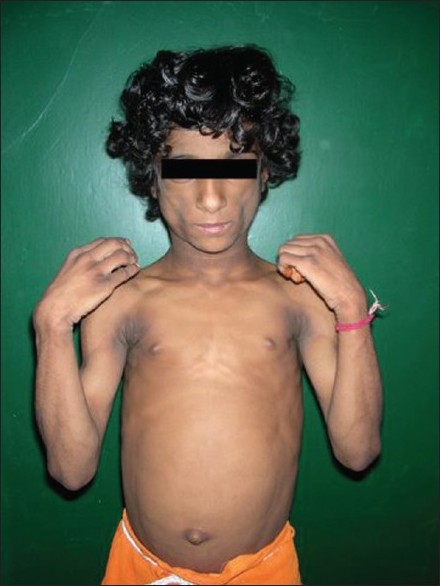 |
| Figure 1: Girl with generalized loss of subcutaneous fat, severe acanthosis nigricans, curly scalp hair, and protuberant abdomen |
Her hemoglobin was 7.9 gm%. Biochemical parameters showed raised SGOT and SGPT (129 and 99 IU/l, respectively), but normal alkaline phosphatase; Hypertriglyceridemia was present (triglyceride 230 mg/dl; normal: 26-123 mg/dl). She had impaired oral glucose tolerance test: the fasting blood sugar was 114 mg/dl (normal: <100 mg/dl) and after 2 hours the blood sugar was 168 mg/dl (normal: <140 mg/dl). Thyroid profile was normal. Ultrasound abdomen showed gross hepatosplenomegaly. CT scan of the abdomen revealed multiple enlarged lymph nodes as well as mural and mucosal prominence in small bowel loops, including in the terminal ileum and ileocecal junction (a feature suggestive of nonspecific enterocolitides). X-rays of the hands revealed periosteal thickening, with osteopenia of the lower ends of the radius and ulna and the small bones of hands [Figure - 2]. Whole-body fat analysis by DEXA scan showed total body fat of 7.3% (normal: 16.5%) and associated generalized osteopenia. Upper gastrointestinal endoscopy revealed maintained villous pattern, with stroma showing mononuclear infiltrate on biopsy, transglutaminase IgA level of 172 U/ml, and antiendomysial IgA antibody level of 19.2 U/ml. In view of these features she was diagnosed to have congenital lipodystrophy syndrome of the Berardinelli-Seip type with associated celiac disease. She was maintained on a gluten-free diet.
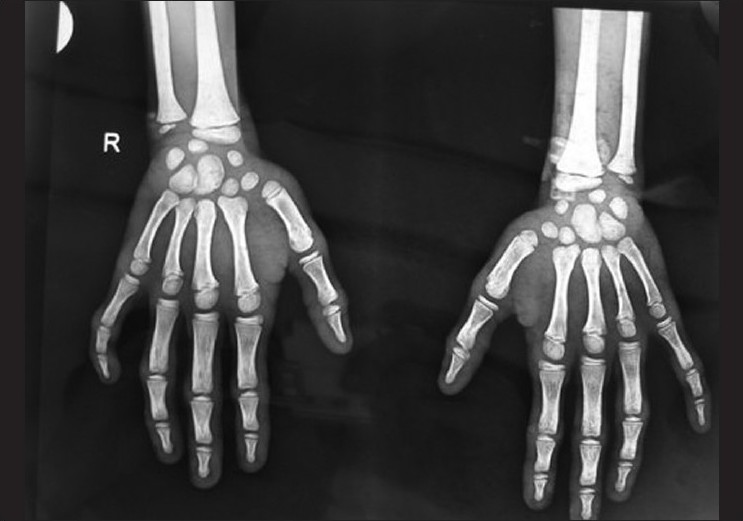 |
| Figure 2: X-ray of the hands showing periosteal thickening with osteopenia of lower ends of the radius and ulna and the small bones of the hands |
Discussion
Generalized congenital lipoatrophy or BSCL was first described in 1954 by Berardinelli in a 2-year-old Brazilian boy and later by Seip in three patients. [4] The defect in BSCL is in the 1-acylglycerol-3-phosphate-O-acyltransferase-2 (AGPAT2) gene on chromosome 9q34 in the type 1 variant and in the BSCL2 gene on chromosome 11q13 in the type 2 variant. It is a rare disorder. There are approximately 250 cases registered all over the world, with greater frequency reported in some ethnic groups, mainly in Latin Americans and Arabians (individuals of Portuguese and Norwegian ancestry). [5]
In BSCL, fat is virtually absent, not just in the subcutaneous area but all over the body. The genetic alteration results in a defect in the development of metabolically active adipose tissue, with decrease in the size and number of adipocytes. Patients with type 1 disease lose all metabolically active adipose tissue present in most subcutaneous areas, in intra-abdominal and intrathoracic regions, and in bone marrow; however, they have well-preserved mechanical adipose tissue over the palms and soles, under the scalp, and in the retro-orbital and periarticular regions. In type 2 disease, patients lose both types of adipose tissue. [6] Muscular hypertrophy is present from birth, with increase in formation of muscle glycogen and creatinine. Skeletal maturation occurs quickly during the first 6-8 years of life [7] and there may be enlarged joints of hands and feet. [8] Acanthosis nigricans is present in almost all cases. It is usually symmetric and involves the flexures, palms, and soles. This entity is closely associated with insulin resistance. [9]
BSCL is associated with several metabolic alterations, including severe hyperlipidemia and insulin resistance. Diabetes installation occurs generally at around 12 years of age, with development of ketosis-resistant diabetes mellitus. Hypermetabolism is an important feature and is associated with increased appetite; there is ingestion of large amounts of calories and profuse sweating. There may be hyperthermia. The endocrine pancreas has an important role in the development of hyperinsulinemia and diabetes. [4]
Cardiac abnormalities in this disorder include ventricular dysfunction and hypertrophic cardiomyopathy. The cardiomyopathy is correlated with the high plasma insulin levels, which activate the type 1 insulin-like growth factor (IGF1) receptors in the myocardial tissue. [10] Hepatic abnormalities range from abnormal liver function tests to adipose tissue infiltration, hepatomegaly, hepatic steatosis, and cirrhosis. Splenomegaly and phlebomegaly (mostly in the extremities) is a feature. [7] There is also slight increase in the size of the external genitalia - the penis and the clitoris - since childhood. [4] Women may have hirsutism, oligomenorrhea, and polycystic ovarian disease (PCOD). [11]
Three major criteria or two major plus two or more minor criteria make a diagnosis of BSCL likely. Major criteria include lipoatrophy affecting the trunk, limbs, and face; acromegaloid features; hepatomegaly; elevated serum triglycerides; and insulin resistance. Minor criteria include hypertrophic cardiomyopathy, psychomotor retardation, hirsutism, precocious puberty in females, bone cysts, and phlebomegaly.
Our patient presented with generalized loss of subcutaneous fat, prominent pectoral and thigh muscles, severe acanthosis nigricans, generalized hyperpigmentation, prominent subcutaneous veins, osteopenia, protuberant abdomen, enlarged clitoris, and hepatosplenomegaly; she had raised transaminases, impaired glucose tolerance test, and hypertriglyceridemia. She also had concomitant celiac disease. This association of celiac disease with generalized lipodystrophy has not been documented earlier, although there is a report of partial lipodystrophy with celiac disease, where the patient had deficiencies of serum IgA and C3 complement, subclinical dermatitis herpetiformis (confirmed by skin biopsy), and persistent severe wasting of the face, upper limbs, and trunk, with sparing of the pelvis and lower limbs. [12]
BSCL should be differentiated from other congenital generalized lipodystrophy syndromes like mandibuloacral dysplasia, Hutchinson-Gilford progeria syndrome, and Werner syndrome (pangeria), which are all premature ageing syndromes.
Patients with BSCL should have multidisciplinary follow-up. Metformin is the drug of choice, since it reduces appetite and improves the symptoms of PCOD and hepatic steatosis. Administration of leptin to children without diabetes - but with insulin resistance, hepatic steatosis, and hypertriglyceridemia - for 4 months has been shown to cause reduction in triglyceride levels, increase in sensitivity to insulin, and reduction in liver volume. BSCL patients should consume a low-fat diet, with low saturated transfats and cholesterol. This should be combined with increase in physical activity. [7]
| 1. |
Berardinelli W. An undiagnosed endocrinometabolic syndrome: Report of two cases. J Clin Endocrinol Metab 1954;14:193-204.
[Google Scholar]
|
| 2. |
Garg A. Acquired and Inherited Lipodystrophies. N Engl J Med 2004;350:1220-34.
[Google Scholar]
|
| 3. |
Hegele RA, Joy TR, Al-Attar SA, Rutt BK. Lipodystrophies: Windows on adipose biology and metabolism. J Lipid Res 2007;48:1433-44.
[Google Scholar]
|
| 4. |
Seip M, Trygstad O. Generalized lipodystrophy, congenital and acquired (lipoatrophy). Acta Paediatr 1996;l413:2-28.
[Google Scholar]
|
| 5. |
Daher E, Silva Júnior G, Benevides V, Mendonça P, Bezerra H, Silva A, et al. Berardinelli syndrome. A case report with fatal outcome. Invest Clin 2008;49:251-5.
[Google Scholar]
|
| 6. |
Garg A, Fleckenstein J, Peshock R, Grundy S. Peculiar distribution of adipose tissue in patients with Congenital Generalized Lipodystrophy. J Clin Endocrinol Metab 1992;75:358-61.
[Google Scholar]
|
| 7. |
Gomes KB, Pardini VC, Fernandes AP. Clinical and molecular aspects of Berardinelli Seip Congenital Lipodystrophy (BSCL). Clin Chim Acta 2009;402:1-6.
[Google Scholar]
|
| 8. |
Darmasdt GL, Sidbury R. The Skin. In: Behrman RE, Kliegman RM, Jenson HB, editors. Nelson Textbook of Paediatrics. 17 th ed. Philadelphia: W B Saunders; 2003. p. 2153-250.
[Google Scholar]
|
| 9. |
Rendon M, Cruz PD, Sontheimer R, Bergstresser P. Acanthosis nigricans: A cutaneous marker of tissue resistance to insulin. J Am Acad Dermatol 1989;21:461-9.
[Google Scholar]
|
| 10. |
Bhayana S, Siu V, Joubert G, Clarson C, Cao H, Hegele R. Cardiomyopathy in congenital complete lipodystrophy. Clin Genet 2002;61:283-7.
[Google Scholar]
|
| 11. |
Huseman CA, Johanson AJ, Varma MM, Blizzard RM. Congenital lipodystrophy. II. Association with polycystic ovarian disease. J Pediatr 1979;95:72-4.
[Google Scholar]
|
| 12. |
O'Mahony D, O'Mahony S, Whelton MJ, McKiernan J. Partial lipodystrophy in coeliac disease. Gut 1990;31:717-8.
[Google Scholar]
|
Fulltext Views
3,818
PDF downloads
2,194





![[Figure - 1]](#fig_ijdvl_2011_77_3_402_79740_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2011_77_3_402_79740_f2.jpg){kind=link}