Translate this page into:
Cutaneous angiosarcoma in a patient with systemic sclerosis: First case from India
2 Department of Pathology, Lady Hardinge Medical College, New Delhi, India
Correspondence Address:
Shiwangi Rana
Flat 901, Tower 2, Sector 52, Gurgaon - 124 001, Haryana
India
| How to cite this article: Mendiratta V, Rana S, Manickavasagam S, Nangia A, Chander R. Cutaneous angiosarcoma in a patient with systemic sclerosis: First case from India. Indian J Dermatol Venereol Leprol 2018;84:214-217 |
Sir,
Cutaneous angiosarcoma is a rare malignant vascular tumor, which has been reported seldom with systemic sclerosis.[1],[2],[3],[4] We are reporting a case of cutaneous angiosarcoma (face) occurring over the sclerodermoid skin in a patient with systemic sclerosis. To the best of our knowledge, this has not been previously reported from the Indian subcontinent.
A 56-year-old female, already diagnosed with systemic sclerosis and interstitial lung disease since the last 6 years, presented with a rapidly progressing, painful, bluish red and hard swelling over the left side of the forehead since 1 month. Cutaneous examination revealed generalized sclerosis and a large well-defined, bluish-red, woody hard, tender plaque (7 × 5 cm) with multiple hemorrrhagic vesicles and bullae was noted over the left temporoparietal area with extensions over the forehead, zygomatic, and maxillary areas [Figure - 1], [Figure - 2], [Figure - 3], [Figure - 4]. Differential diagnoses of angiosarcoma and mycetoma were considered. Investigations showed elevated erythrocyte sedimentation rate (34 mm/h), positive antinuclear antibodies (3+), Scl-70 (3+) and SS-A antibodies (+). Computed tomography scan of the head showed edema in the subcutaneous plane.
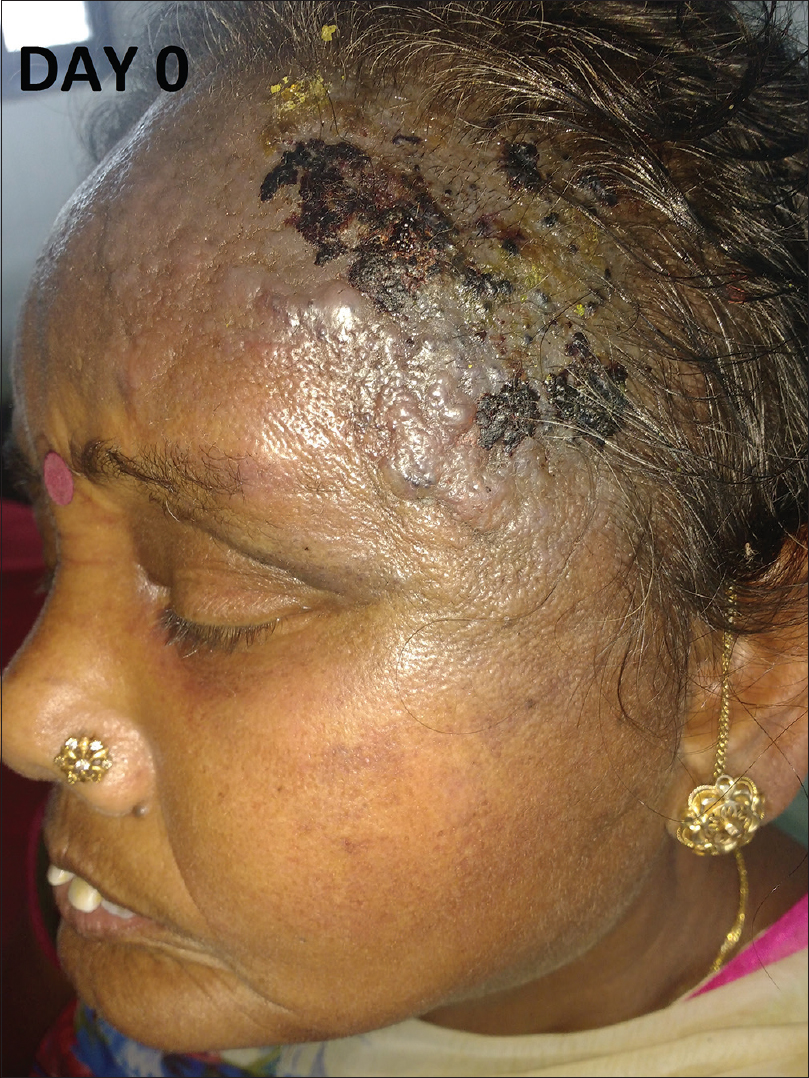 |
| Figure 1: Bluish red plaque with hemorrhagic blister over the sclerodermoid skin of the scalp and forehead |
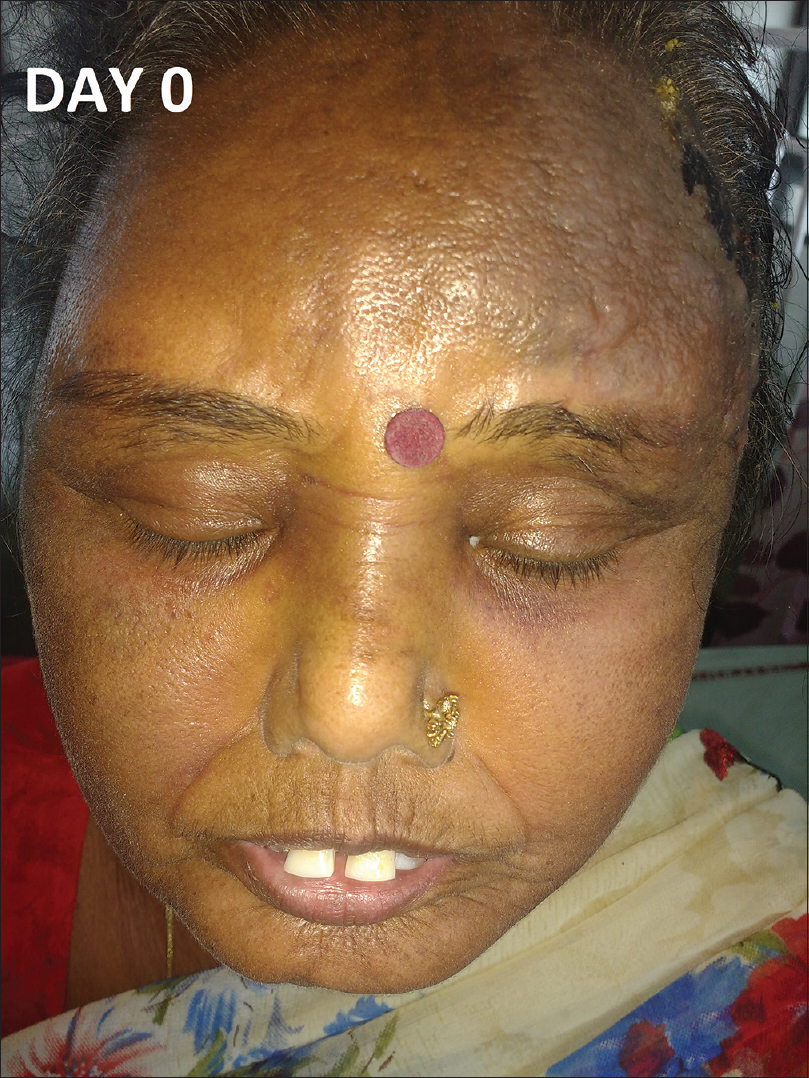 |
| Figure 2: Bluish red plaque with hemorrhagic blister over the sclerodermoid skin of the scalp and forehead |
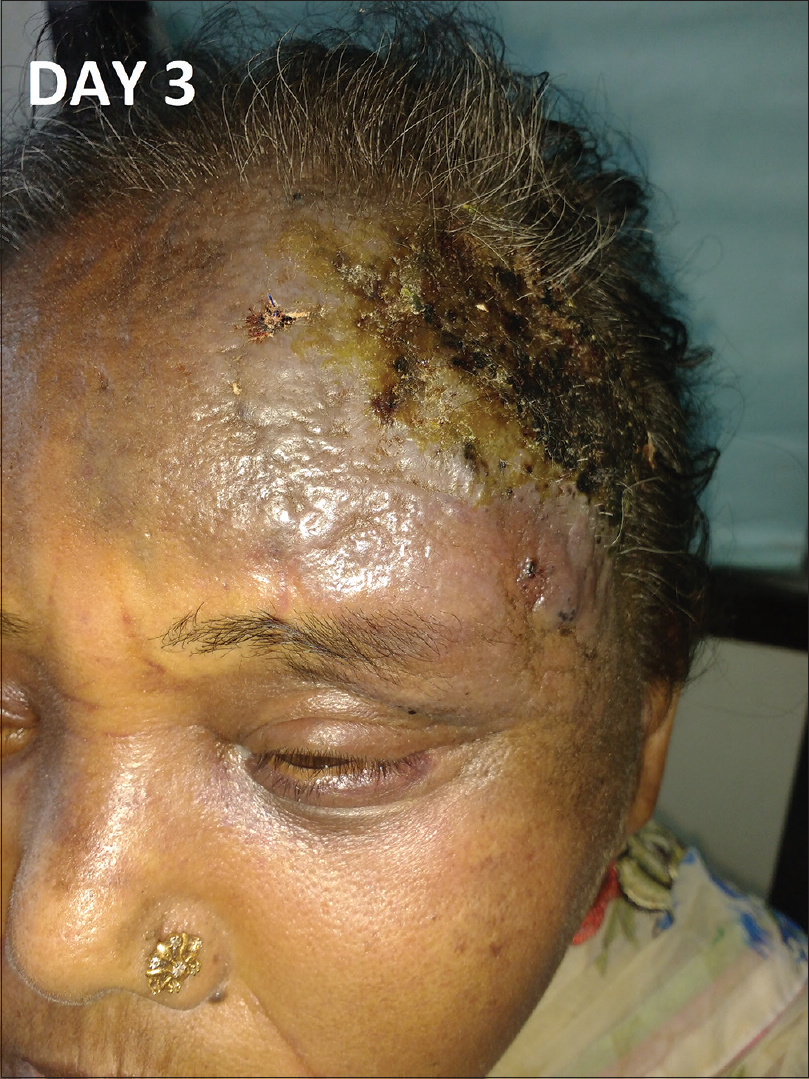 |
| Figure 3: Rapid extension of the lesion with crusting and erosion over the surface on day 3 of admission |
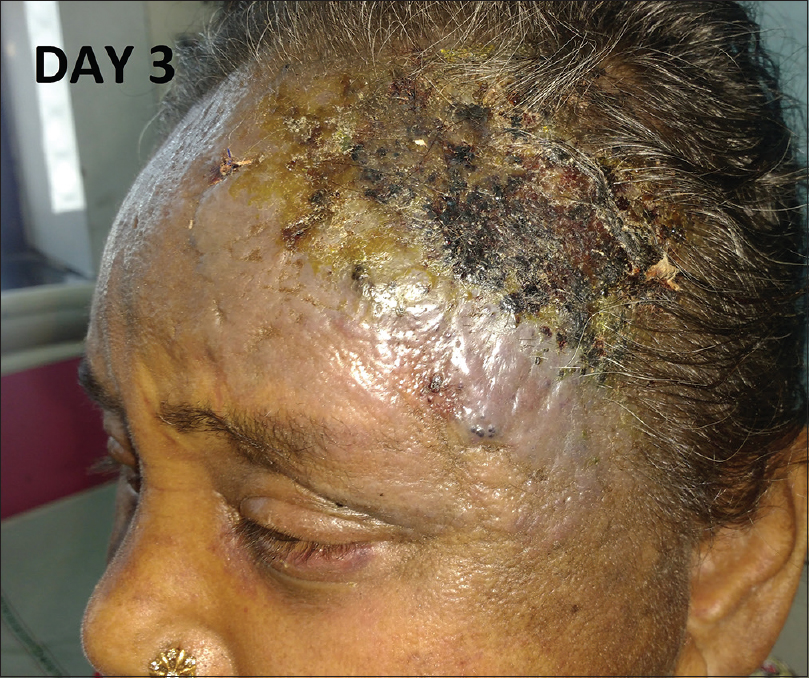 |
| Figure 4: Rapid extension of the lesion with crusting and erosion over the surface on day 3 of admission |
Fine needle aspiration cytology from the plaque showed clusters of malignant cells with mitotic figures, suggestive of a malignant pathology. Incisional skin biopsy from the same site revealed a few branching vascular channels in the dermis, lined by a single row of flat to plump endothelial cells [Figure - 5]. The individual cells had scant eosinophilic cytoplasm with enlarged round to oval nucleus [Figure - 6]. Atypical cells were positive for CD34 and CD31 [Figure - 7] and [Figure - 8], strongly positive for Ki67, and negative for leukocyte common antigen (LCA). A final diagnosis of cutaneous angiosarcoma was made. No improvement was seen even after six cycles of chemotherapy.
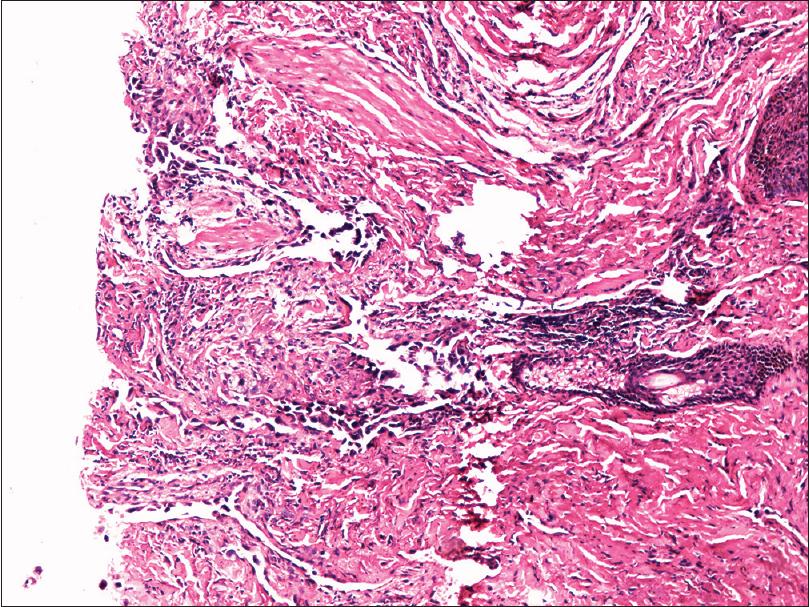 |
| Figure 5: Dermis showing irregular anastomosing vascular channels (H and E, ×100) |
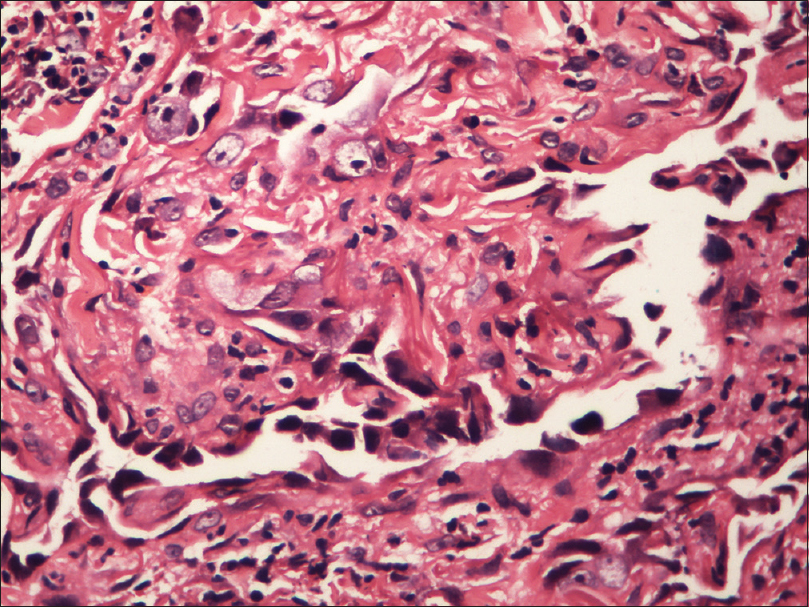 |
| Figure 6: Magnified view of vascular channels in the dermis lined by endothelial cells (H and E, ×100) |
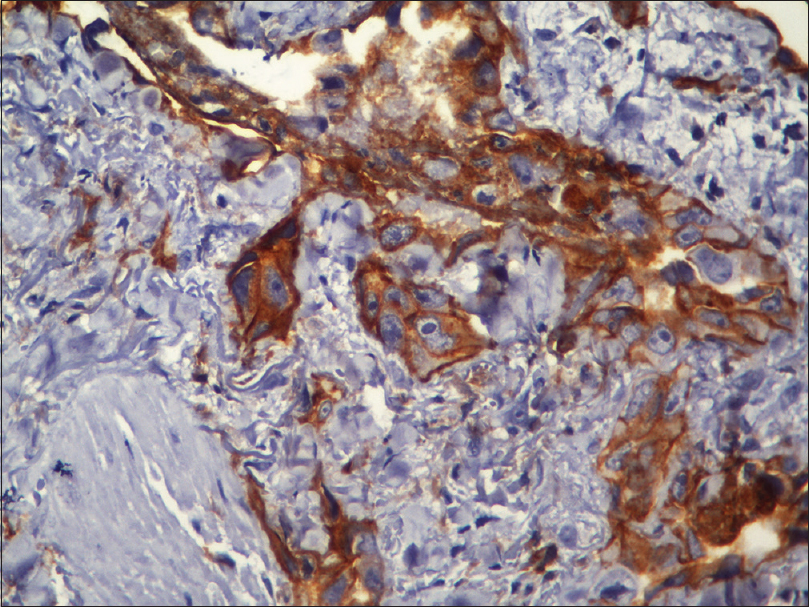 |
| Figure 7: Immunohistochemistry showing CD31 positive cells (immunohistochemistry, ×400) |
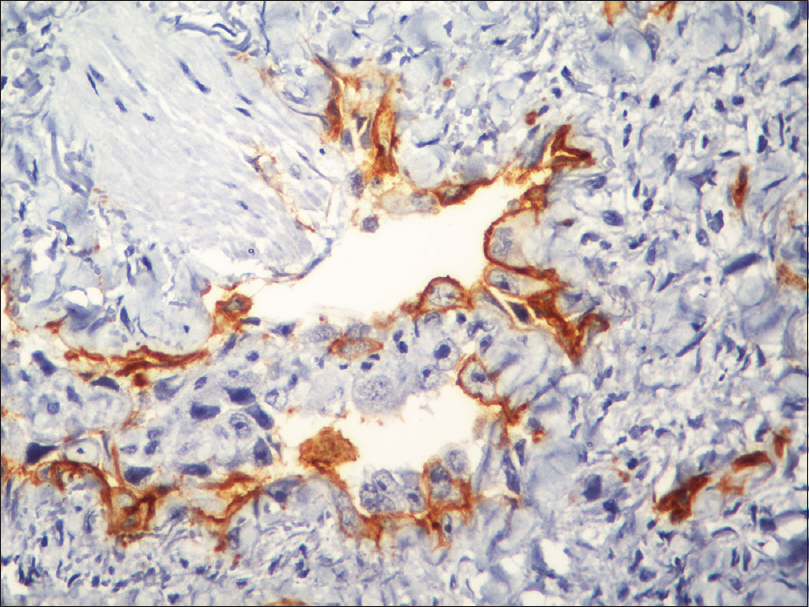 |
| Figure 8: Immunohistochemistry showing CD34 positive cells (immunohistochemistry, ×400) |
Angiosarcoma is a rare, malignant vascular tumor that is localized to the skin or superficial soft tissue. It is known to occur in three different clinical settings – idiopathic cutaneous angiosarcoma of the head and neck (most common), angiosarcoma complicating lymphedema and post-irradiation angiosarcoma. The cancerous lesion rapidly progresses and has a fulminant course.
Histologically, low-grade angiosarcoma is composed of irregular vascular channels dissecting dermal collagen, lined by atypical endothelial cells. In a few cases, tumor can show high grade differentiation with vascular anastomosing channels lined by atypical endothelium, resembling high-grade sarcoma and carcinoma. Immunohistochemistry with endothelial specific antibodies (Ulex europaeus agglutinin, antibody to factor VIII related, and CD31 and 34 antigens) helps in confirming the diagnosis.
The exact mechanism responsible for tumorigenesis in systemic sclerosis patients is still unknown. Multiple mechanisms have been proposed; such as endothelial cell damage through ischemia, growth modulatory mediators released from inflammatory cells and platelets, genetic susceptibility, decline in the number of natural killer T cells, therapeutically induced immunosuppression and vascular endothelial growth factor (VEGF) overexpression.[5]
Shah et al.[6] investigated 23 systemic sclerosis patients with malignancy and concluded that patients with RNA polymerase III antibodies had a uniquely close temporal relationship with cancer diagnosis and systemic sclerosis onset.
The site of angiosarcoma was similar to the earlier reported cases of systemic sclerosis with angiosarcoma [Table - 1]. Levels of anti-RNA polymerase 3 antibodies and VEGF could not be assessed. It would be interesting to evaluate the levels of serum VEGF in systemic sclerosis and to determine its cut-off levels to predict the risk of developing angiosarcoma in the subset of patients who have higher/very high levels of VEGF. Treatment with anti-VEGF monoclonal antibodies (Bevacizumab) in this subset of patients might help in arresting the neoplastic process.

Angiosarcoma can pose diagnostic challenges as it can mimic other benign vascular lesions. Immunohistochemistry and detailed neuroimaging studies can minimize the delay in diagnosis. To the best of our knowledge, this case is the fifth reported case of angiosarcoma in association with Systemic sclerosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understands that her name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Puizina-Ivic N, Bezic J, Marasovic D, Gotovac V, Carija A, Bozic M. Angiosarcoma arising in sclerodermatous skin. Acta Dermatovenerol Alp Pannonica Adriat 2005;14:20-5.
[Google Scholar]
|
| 2. |
Fonder MA, Douglas DK. Angiosarcoma complicating systemic sclerosis: A case report. Cutis 2008;81:468-72.
[Google Scholar]
|
| 3. |
Kubota N, Fujisawa Y, Nakamura Y, Tanaka R, Saito A, Maruyama H. Angiosarcoma of the Scalp in a Patient with System Sclerosis. The Journal of Dermatology 2015:42:102-4.
[Google Scholar]
|
| 4. |
Carter B, Jaworsky C, Fox M. Systemic sclerosis associated angiosarcoma: A case report and review of the literature. Int J Dermatol Clin Res 2016;2(1):14-7.
[Google Scholar]
|
| 5. |
Arbiser JL, Larsson H, Claesson-Welsh L, Bai X, LaMontagne K, Weiss SW, et al. Overexpression of VEGF 121 in immortalized endothelial cells causes conversion to slowly growing angiosarcoma and high level expression of the VEGF receptors VEGFR-1 and VEGFR-2 in vivo. Am J Pathol 2000;156:1469-76.
[Google Scholar]
|
| 6. |
Shah AA, Rosen A, Hummers L, Wigley F, Casciola-Rosen L. Close temporal relationship between onset of cancer and scleroderma in patients with RNA polymerase I/III antibodies. Arthritis Rheum 2010;62:2787-95.
[Google Scholar]
|
Fulltext Views
3,158
PDF downloads
2,714





![[Figure - 1]](#fig_ijdvl_2018_84_2_214_224001_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2018_84_2_214_224001_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2018_84_2_214_224001_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2018_84_2_214_224001_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2018_84_2_214_224001_f5.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2018_84_2_214_224001_f6.jpg){kind=link}
![[Figure - 7]](#fig_ijdvl_2018_84_2_214_224001_f7.jpg){kind=link}
![[Figure - 8]](#fig_ijdvl_2018_84_2_214_224001_f8.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2018_84_2_214_224001_t9.jpg){kind=link}