Translate this page into:
D66H mutation in GJB2 gene in a Chinese family with classical Vohwinkel syndrome
2 Department of Dermatology, Provincial Hospital Affiliated to Shandong University, Jinan, Shandong, China
Correspondence Address:
Li Zhang
Department of Dermatology, Provincial Hospital Affiliated to Shandong University, 324 Jingwuweiqi Road, Jinan, Shandong Province
China
| How to cite this article: Qiu Y, Wang Z, Chen N, Song Y, Wang Z, Zhang L. D66H mutation in GJB2 gene in a Chinese family with classical Vohwinkel syndrome. Indian J Dermatol Venereol Leprol 2012;78:640-642 |
Sir,
Vohwinkel syndrome (VS; OMIM 124500), also known as mutilating palmoplantar keratoderma, is a rare autosomal dominant skin disease first described by Vohwinkel in 1929. VS is characterized by diffuse hyperkeratosis of palms and soles, a honeycomb appearance, starfish-like keratoses, and constriction bands leading to auto-amputation of the digits (pseudo-ainhum). Two candidate genes, LOR and GJB2, were identified by Maestrini et al. in 1996 and 1999, respectively. [1] GJB2 encodes connexin 26 (Cx26), a component of intercellular gap junctions. VS caused by mutation of GJB2 usually involves deafness and is considered to be classical VS. Mutation of the LOR gene (encoding loricrin) causes VS with concomitant ichthyosis and is classified as a variant VS.
Here, we report classical VS caused by D66H mutation in GJB2 in a Chinese family. To our knowledge, this is the first time to suggest that D66H mutation in GJB2 gene is involved in the etiology of Chinese classical VS. The study was approved by the Ethics Committee of the Shandong University and conformed to Declaration of Helsinki Principles.
A four-generation family of 32 individuals, including five affected individuals (one male and four females), was identified in the Shandong province of China. The mode of inheritance was determined to be autosomal dominant [Figure - 1]. All patients had similar clinical symptoms, characteristic of the disease, as previously described [Figure - 2]a-c, and all affected individuals had moderate-to-severe sensorineural hearing loss. The patients were in good general health. No ichthyosis, eye abnormalities, facial dysmorphism, oral leukokeratosis, dental and hair anomalies were found.
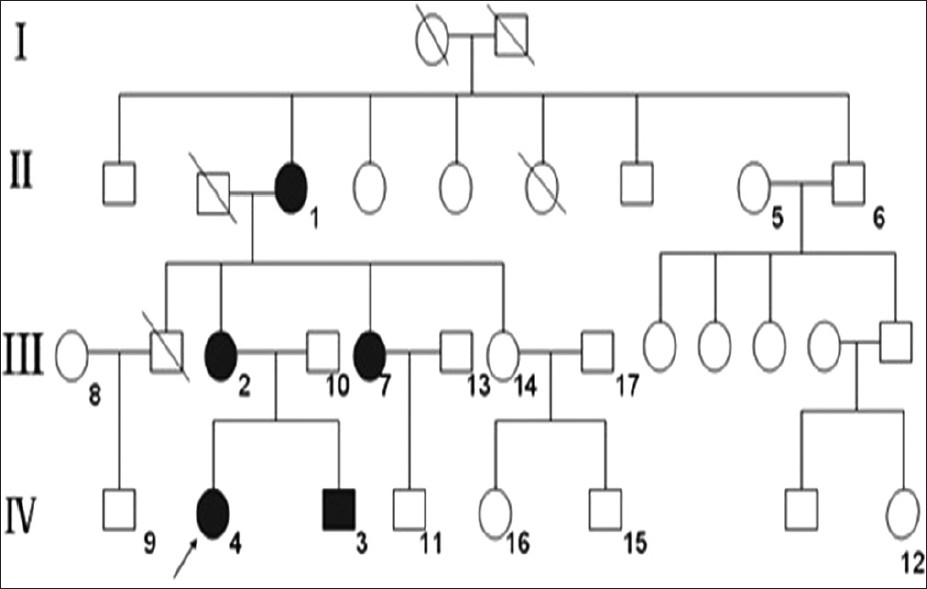 |
| Figure 1: The pedigree of the Chinese family with classical VS showing autosomal dominant inheritance. Arrow indicates proband. I, II, III, IV: Generation numbers. 1-17: Study object |
Our patient had been treated with acitretin 30 mg daily and Chinese herbal drugs whose main function is activating blood circulation and dissipating blood stasis, these medications made some improvement in the condition, significant thinning of the keratoderma and release of the pseudo-ainhums were seen.
After informed consents were obtained, peripheral blood samples were collected from all five affected and seven unaffected family members, as well as 100 healthy controls. DNA was extracted, and the entire coding region of the GJB2 gene was amplified by the polymerase chain reaction (PCR) using the following primers: forward 5´-ACCTGTTTTGGTGAGGTTGTG-3´ and reverse 5´-CCATTGTGGCATCTGGAGTT-3´. PCR products were bidirectionally sequenced using an ABI 3730XL automated sequencer.
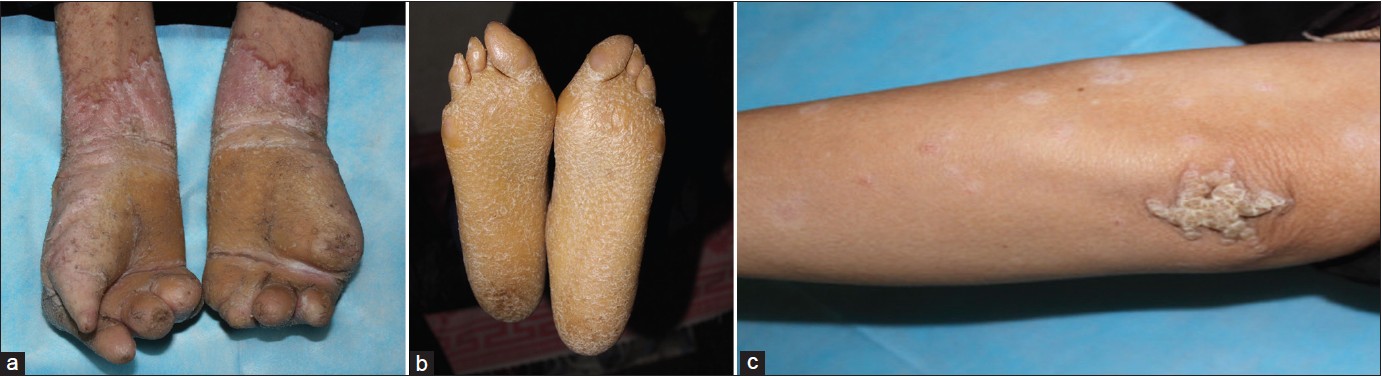 |
| Figure 2: Clinical appearance of the affected individuals studied and genetic analysis of Cx26 (a, b) (II-1) Diffuse hyperkeratosis of palms and soles with a honeycomb appearance, all the fingers and the little toe had auto-amputation (c) (III-2) The extensor aspect of right elbow joint shows starfish-like keratoses |
D66H mutation in GJB2 gene, resulting in a histidine to aspartic acid substitution at codon 66 of Cx26 [Figure - 3]a and b, was found in all affected individuals of this family, while none of the 12 unaffected family members or the 100 control individuals possessed this mutation. These results suggest that this locus is not polymorphic in the Chinese Han population.
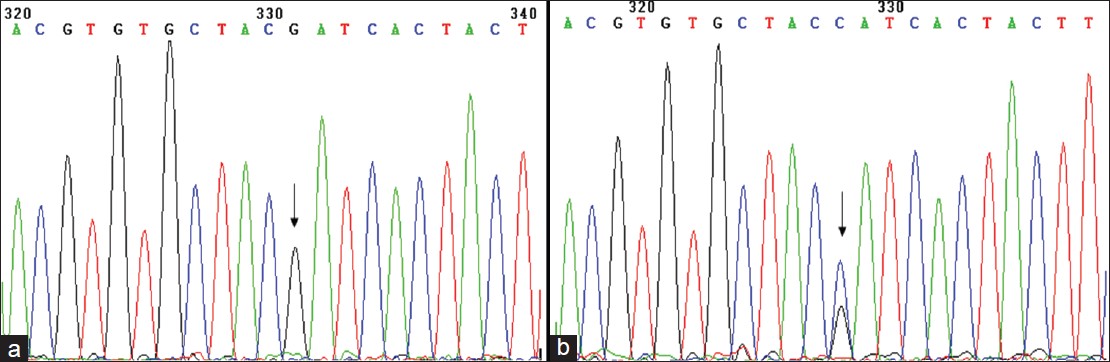 |
| Figure 3: (a) Normal DNA sequence of GJB2 (+ strand) corresponding to codons 63-69 (b) Same region as shown in (II-1) from the affected individual showing heterozygous missense mutation 196 G→ C, predicting amino acid change D66H |
D66H mutation in GJB2 gene was first identified by Maestrini et al. in three unrelated families, which come from different countries (British, Italian and Spanish), presenting with classical VS. [1] Here, we report a D66H mutation in a Chinese pedigree presenting with classical VS. Our results further support the theory that D66H is a mutational ′′hotspot′′ in the GJB2 gene associated with classical VS.
Up until now, four heterozygous missense mutations in the GJB2 gene have been reported to be associated with classical VS: D66H, G130V, G59S and Y65H. [1],[2],[3],[4] Three of these are found in the first extracellular loop (E1), one is located in the second intracellular domain, and all are single amino acid substitutions. The D66H and Y65H mutations can cause almost identical disease phenotypes including hyperkeratosis of palms and soles, starfish-like keratoses, constriction bands of the digits and mild progressive hearing loss. [1,4] Sometimes, the D66H mutation can induce epilepsy, [5] paratrichosis, vitiligo, and so on. The patients carrying the G130V mutation showed profound hearing loss and mild palmoplantar keratoderma, some of them may have no sign of constrictions of the toes or fingers. [2],[6] The G59S mutation can lead to mutilating keratoderma, combined with generalized ichthyosis and complete congenital deafness. Allegedly, this mutation has a particular proneness to skin cancer. [3] Expression of the normal human GJB2 gene produced no abnormalities.
Previous studies suggested that Cx26 (D66H) could inhibit the channel function of wild-type Cx26 and Cx43. [7] The gap junction plaques formed by Cx26 (D66H) would most likely involve interference with trafficking of wild-type Cx30 in the K10Cx26 (D66H) mouse epidermis. Recent data suggested that Cx26 (D66H) could inhibit Cx43 channel activity when expressed in Xenopus oocytes. However, it did not appear to disrupt membrane localization of Cx43 in C33a cells or in the epidermis. [8] Studies suggested that homozygous loss of either Cx26 or Cx30 expressed in epidermis could compensate for each other. [7],[8]
Experimental evidence confirmed that D66H and Y65H form residual gap junction plaques. Parachute assay showed reduced dye transfer as a result of these mutations. [1],[4],[5] Therefore, it is possible that these genetic mutations result in transport defects that lead to classical VS. Moreover, connexins have been demonstrated to be involved in the regulation of growth and differentiation of the epidermis, and their mutation may be associated with the pathogenesis of VS. However, the precise mechanisms are still unclear.
In summary, evidence from previous and current research leads us to believe that GJB2 gene mutation plays an important role in the pathogenesis of VS in humans. Further investigation will focus on determination of the function of mutant GJB2 and elucidation of the role of D66H mutation in VS.
Acknowledgement
We are very grateful to the family members and the 100 healthy controls for their participation in the study. This work was supported by the National Natural Science Foundation of China (N0.81171492) and Shandong Province Science and Technology Development Planning (2010GSF10812 and 2011GSF11847).
| 1. |
Maestrini E, Korge BP, Ocaña-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, et al. A missense mutation in connexin 26, D66H, causes mutilating keratoderma with sensorineural deafness (Vohwinkel's syndrome) in three unrelated families. Hum Mol Genet 1999;8:1237-43.
[Google Scholar]
|
| 2. |
Snoeckx RL, Hassan DM, Kamal NM, Van Den Bogaert K, Van Camp G. Mutation analysis of the GJB2 (connexin 26) gene in Egypt. Hum Mutat 2005;26:60-1.
[Google Scholar]
|
| 3. |
Bondeson ML, Nyström AM, Gunnarsson U, Vahlquist A. Connexin 26 (GJB2) mutations in two Swedish patients with atypical Vohwinkel (mutilating keratoderma plus deafness) and KID syndrome both extensively treated with acitretin. Acta Derm Venereol 2006;86:503-8.
[Google Scholar]
|
| 4. |
de Zwart-Storm EA, van Geel M, Veysey E, Burge S, Cooper S, Steijlen PM, et al. A novel missense mutation in GJB2, p.Tyr65His, causes severe Vohwinkel syndrome. Br J Dermatol 2011;164:197-9.
[Google Scholar]
|
| 5. |
Serrano Castro PJ, Naranjo Fernandez C, Quiroga Subirana P, Payan Ortiz M. Vohwinkel Syndrome secondary to missense mutation D66H in GJB2 gene (connexin 26) can include epileptic manifestations. Seizure 2010;19:129-31.
[Google Scholar]
|
| 6. |
Lossa S, Chinetti V, Auletta G, Laria C, De Luca M, Rienzo M, et al. New evidence for the correlation of the p. G130V mutation in the GJB2 gene and syndromic hearing loss with palmoplantar keratoderma. Am J Med Genet A 2009;149:685-8.
[Google Scholar]
|
| 7. |
Rouan F, White TW, Brown N, Taylor AM, Lucke TW, Paul DL, et al. Trans-dominant inhibition of connexin-43 by mutant connexin-26: Implications for dominant connexin disorders affecting epidermal differentiation. J Cell Sci 2001;114:2105-13.
[Google Scholar]
|
| 8. |
Bakirtzis G, Choudhry R, Aasen T, Shore L, Brown K, Bryson S, et al. Targeted epidermal expression of mutant Connexin 26 (D66H) mimics true Vohwinkel syndrome and provides a model for the pathogenesis of dominant connexin disorders. Hum Mol Genet 2003;12:1737-44.
[Google Scholar]
|
Fulltext Views
3,105
PDF downloads
2,042





![[Figure - 1]](#fig_ijdvl_2012_78_5_640_100595_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_5_640_100595_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_5_640_100595_f3.jpg){kind=link}