Translate this page into:
De Sanctis-Cacchione syndrome
2 Department of Pediatrics, Lok Nayak Hospital, New Delhi, India
Correspondence Address:
Sumit Mehndiratta
Department of Pediatrics, Lok Nayak Hospital, New Delhi - 110 002
India
| How to cite this article: Mittal H, Mehndiratta S, Kaushik JS, Godbole T. De Sanctis-Cacchione syndrome. Indian J Dermatol Venereol Leprol 2013;79:849 |
Sir,
De Sanctis-Cacchione syndrome (DSC) (MIM #278800) is a rare autosomal recessive disorder with a gene map locus of 10q11, originally described as "xerodermic idiocy." [1] DSC is characterized by cutaneous photosensitivity, microcephaly, mental retardation, short stature, hypogonadism, spasticity, peripheral neuropathy and sensorineural deafness. [2] In most of the cases in the literature, hypogonadism has been described, but we report a case of DSC with intact gonadal functions.
A 10-year-old boy presented with multiple episodes of myoclonic seizures and progressive, rapid loss of milestones, cognition, vision and hearing for the past 6 months. There was a history of persistent drooling of saliva, nasal regurgitation of feeds with episodes of choking and coughing with attempted solid feeds. The child was confined to bed and gave no indications for thirst, hunger, bladder or bowel needs. There was no response to auditory and visual stimuli. Past history suggested dry and scaly skin and photosensitivity (since infancy) and developmental delay (developmental quotient 0.33; revised Denver Developmental Scale II). The child was a third in birth order, product of a third degree consanguineous marriage with high maternal (40 years) and paternal (45 years) age, born at term with an uneventful neonatal period. Other siblings were unaffected and there was no family history of photosensitivity, seizures or mental retardation. On examination, the child was emaciated with a weight of 15 kg (<3 rd percentile), length of 122 cm (<3 rd percentile), body mass index of 10.1 (<3 rd centile) and head circumference of 45 cm (<3 rd percentile). Facial features were suggestive of "Old man look" with an elongated face, micrognathia, pinched nose and crowded caries teeth.The skin lesions were noted over the face, neck, upper chest and exposed parts of limbs and consisted of scaling, erythema, crusting with patches of hyperpigmentation [Figure - 1]. He had fixed contractures of the ankle, knee, elbow and wrist joints [Figure - 2]. The genitals, including the penis and testes, were normal with a stretched penile length of 4.5 cm (−2.5 standard deviation = 3.7 cm) and Tanner′s sexual maturity rating (SMR) of stage 2. Examination of the central nervous system revealed that the child had spontaneous eye opening without focusing, motor response of localization to pain and incomprehensible vocal response. Fundus examination revealed the presence of salt and pepper retinopathy. Brainstem evoked auditory response revealed severe sensory-neural hearing loss. Pharyngeal reflex (gag reflex) was weak suggestive of bulbar involvement. On examination, 3 rd , 4 th , 5 th , 6 th , 7 th and 12 th cranial nerves were normal. Motor system examination showed a decrease in muscle bulk, increased tone, reduced power (1/5) and contractures at knee, ankle, elbow and wrist joints. The elicitation of deep tendon reflexes were masked by fixed contractures. There were no abnormal movements and no cerebellar or meningeal signs. Examination of the abdominal, respiratory and cardiovascular systems revealed no other abnormalities. Laboratory investigations showed a normal hemoglobin (Hb 12.1 g/dl), serum creatinine (0.8 g/dl), serum albumin (3.5 g/dl), alanine aminotransferase (24 IU/L) and alkaline phosphatase (120 IU/L). Skeletal survey showed a bone age of 11-12 years. Contrast-enhanced computed tomography (brain) revealed dilated lateral ventricles, atrophy of cerebral and cerebellar cortex and hydrocephalus (ex-vacuo) with no intracranial calcifications [Figure - 3] and [Figure - 4]. Nerve conduction velocity of upper and lower limb suggested axonal neuropathy. Electromyography revealed a decline in motor unit recruitment and other nonspecific changes. Based on the constellation of above features, a diagnosis of DSC was established.
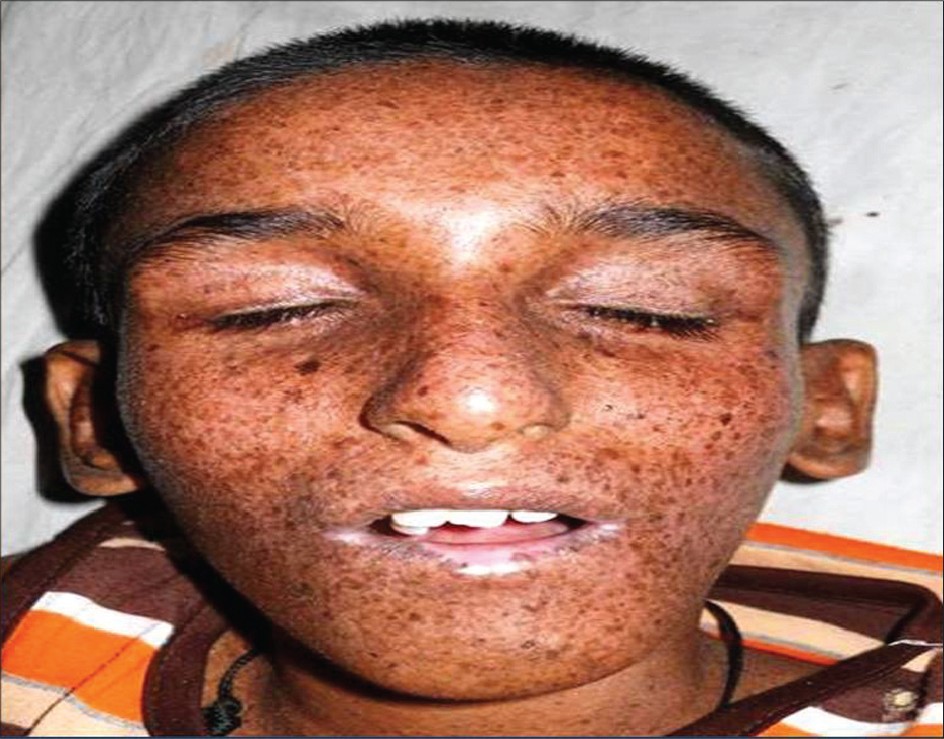 |
| Figure 1: Elongated face with erythema, scaling and crusted lesions |
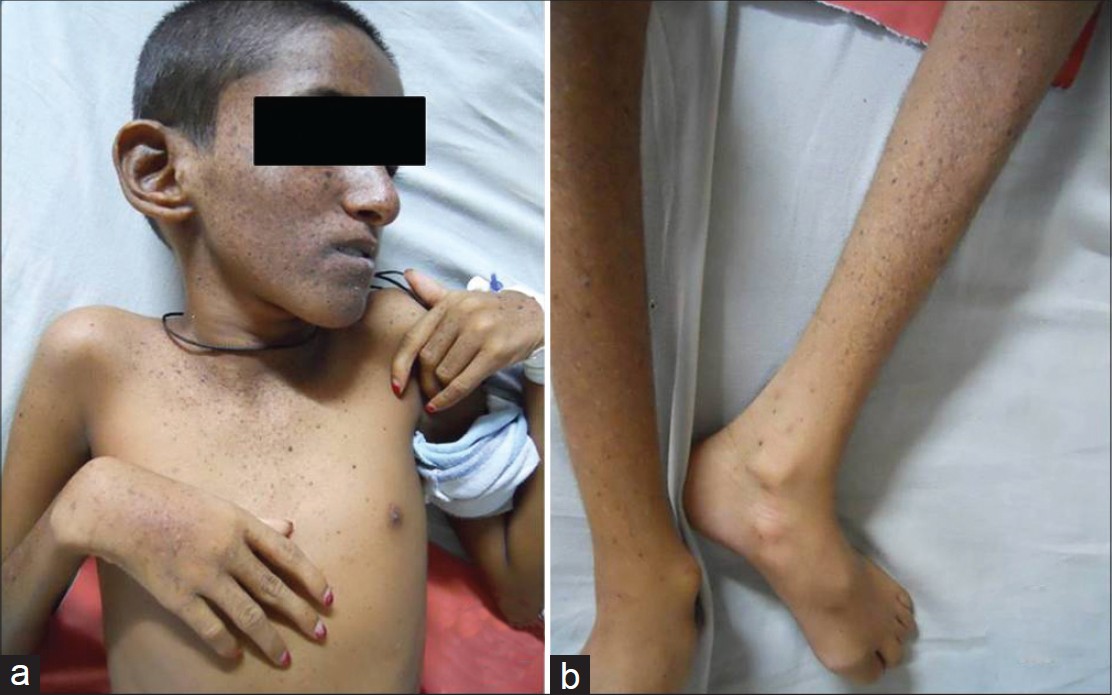 |
| Figure 2: (a and b) Contractures at elbow, wrist, knee and ankle along with lesions on upper chest, hands and legs |
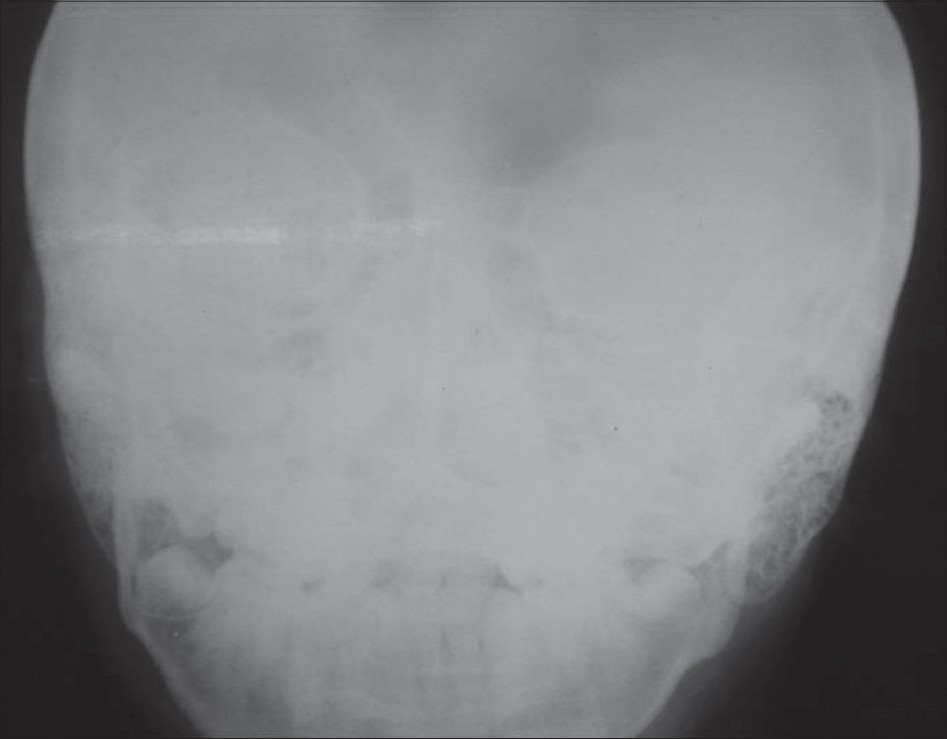 |
| Figure 3: X-ray of skull Anterio-Posterior view showing prominent mastoids and no intracranial calcifi cations |
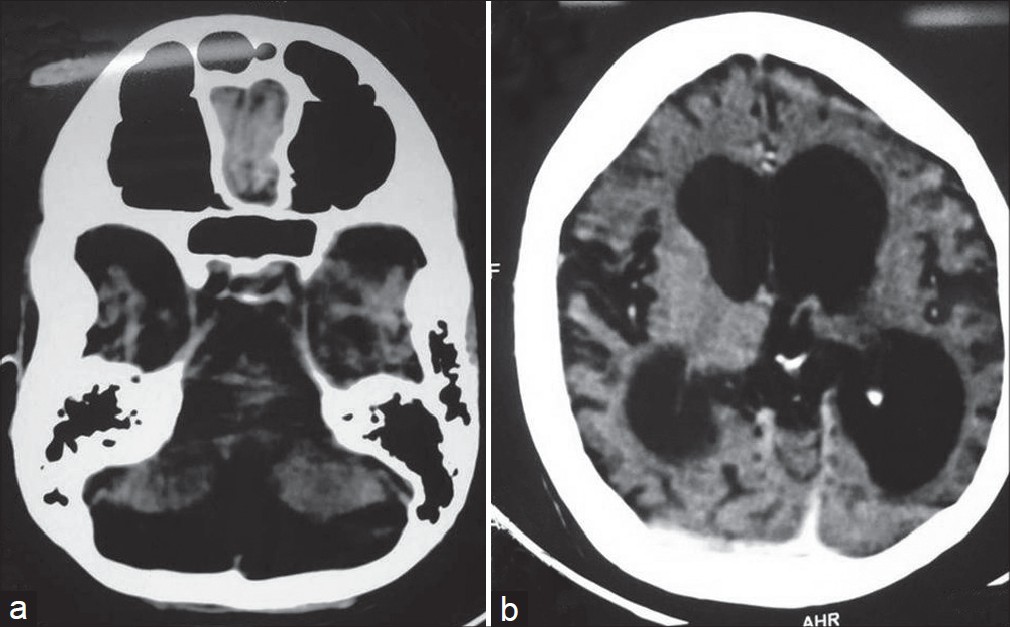 |
| Figure 4: (a and b) Computed tomography scan of patient showing cortical and cerebellar atrophy with ex-vacuo hydrocephalus |
The child was advised protection from sun exposure by the use of protective clothing, eyeglasses, broad spectrum sunscreens containing para-aminobenzoic acid esters and skin emollients. Seizures were controlled with sodium valproate (25 mg/kg) and lamotrigine mg/kg (2.5 mg/kg/day). Physiotherapy was initiated to prevent the worsening of contractures. Child was on regular follow-up for next 6 months. Seizures were controlled with no further progression of neurological illness.
Xeroderma pigmentosa (XP) results from defects in the excision repair of ultraviolet (UV) induced deoxyribonucleic acid (DNA) damage. Defects in nucleotide excision repair (NER) results in inherited neurocutaneous syndromes such as XP, Cockayne syndrome and trichothiodystrophy. There is a complex relationship between NER mutations and clinical features. Seven xeroderma pigmentosum genes, XPA through XPG, have been identified. Conversely, different mutations in one gene can lead to different clinical disorders with variation in systemic involvement. [2],[3] The gene locus implicated in DSC is ERCC6, which is located at 10q11, which is also called Cockayne syndrome-B gene. [4]
Among the variants of XP, patients with DSC have the greatest UV sensitivity and the poorest DNA repair. The neurological manifestations in XP have been hypothesized to be secondary to defective DNA repair in the neuronal cells resulting in neuronal death or dysfunction. Although neurological symptoms appear in the first two decades of life, late presentation in adults has also been reported. [5] The common neurological manifestations of DSC are progressive neurological deterioration, peripheral neuropathy and sensorineural deafness observed in 18% of cases. [6] Other manifestations include microcephaly, choreoathetosis, cerebellar and extra pyramidal syndromes. [7] The late onset of neurological complaints and the relative preservation of gonads and sexual maturity in our case depict sparing of cells from UV induced damage. Early intervention and appropriate protection from sunlight started early in the illness could have a significant impact in delaying the onset of neurological symptoms. There are very few cases reported with complete manifestations of this syndrome. However, many patients have one or more of its neurological features. The features that favored a diagnosis of DSC in our case were mental retardation, microcephaly, cutaneous photosensitivity, stunting, spasticity, contractures, sensorineural deafness and peripheral neuropathy. However, unlike the classical DSC, our child had normal gonads and sexual maturity staging at the time of presentation. In the absence of cataract, optic atrophy, skeletal dysplasia and basal ganglia calcifications, Cockayne syndrome was ruled out.
Diagnosis of DSC is mainly based on clinical features. Chromosomal breakage studies, complementation studies and gene sequencing to identify the specific gene complementation group are supportive. The mainstay of treatment is adequate sun-protection and regular follow up for the early detection of cutaneous malignancies. This condition is not curable and lesser than 40% patients with severe manifestations survive beyond the second decade of life. A multidisciplinary approach, involving a Neurologist and an Ophthalmologist, is required to deal with the associated conditions. A novel approach to photo protection is to repair DNA damage after UV exposure by the delivery of a DNA repair enzyme into the skin by means of specially engineered liposomes. T4 endonuclease V has been shown to repair cyclobutane pyrimidine dimers resulting from DNA damage. [8] Gene therapy for XP is still in a theoretical and experimental stage. The earlier onset of disease is associated with a poorer prognosis. Parental counseling plays a significant role in the reduction of future recurrence as prenatal diagnosis is possible by amniocentesis or chorionic villous sampling.
| 1. |
De Sanctis C, Cacchione A. L'idiozia xerodermica. Riv Sper Freniatr Med Leg Alien Ment 1932;56:269-92.
[Google Scholar]
|
| 2. |
Mishra OP, Tripathi AM, Katiyar GP. DeSanctis-Cacchione syndrome. Indian J Pediatr 1997;64:269-72.
[Google Scholar]
|
| 3. |
Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007;145:1388-96.
[Google Scholar]
|
| 4. |
Colella S, Nardo T, Botta E, Lehmann AR, Stefanini M. Identical mutations in the CSB gene associated with either Cockayne syndrome or the DeSanctis-cacchione variant of xeroderma pigmentosum. Hum Mol Genet 2000;9:1171-5.
[Google Scholar]
|
| 5. |
Robbins JH, Kraemer KH, Merchant SN, Brumback RA. Adult-onset xeroderma pigmentosum neurological disease - Observations in an autopsy case. Clin Neuropathol 2002;21:18-23.
[Google Scholar]
|
| 6. |
Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol 1987;123:241-50.
[Google Scholar]
|
| 7. |
El Fékih N, Fredj M, Aounallah-Skhiri H, Fazaa B, Zghal M, Ridha Kamoun M. Neurological abnormalities in xeroderma pigmentosum. Rev Neurol (Paris) 2009;165:967-70.
[Google Scholar]
|
| 8. |
Yarosh DB, O'Connor A, Alas L, Potten C, Wolf P. Photoprotection by topical DNA repair enzymes: Molecular correlates of clinical studies. Photochem Photobiol 1999;69:136-40.
[Google Scholar]
|
Fulltext Views
3,215
PDF downloads
3,181





![[Figure - 1]](#fig_ijdvl_2013_79_6_849_120760_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_6_849_120760_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2013_79_6_849_120760_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2013_79_6_849_120760_f4.jpg){kind=link}