Translate this page into:
Dorfman-Chanarin syndrome
2 Department of Pathology, University College of Medical Sciences and Guru Teg Bahadur Hospital, Delhi, India
Correspondence Address:
Vijay Gandhi
A-242, Surya Nagar, Ghaziabad - 201 011, UP
India
| How to cite this article: Gandhi V, Aggarwal P, Dhawan J, Singh UR, Bhattacharya S N. Dorfman-Chanarin syndrome. Indian J Dermatol Venereol Leprol 2007;73:36-39 |
Abstract
A four-year-old girl was brought to the dermatology outpatient department with scaling all over the body since birth. She had history of episodic vomiting and abdominal distension. A dermatological diagnosis of lamellar ichthyosis was made. Abdominal examination revealed a nontender hepatomegaly, fatty liver on ultrasonography and deranged liver function tests. Peripheral blood smear showed lipid vacuoles in the granulocytes consistent with Jordans' anomaly. Similar lipid vacuoles were seen in the basal layer in skin biopsy. An inflammatory infiltrate, moderate fibrosis in the portal tract and diffuse severe fatty change in hepatocytes were seen in liver biopsy. The patient was diagnosed as a case of Dorfman-Chanarin syndrome.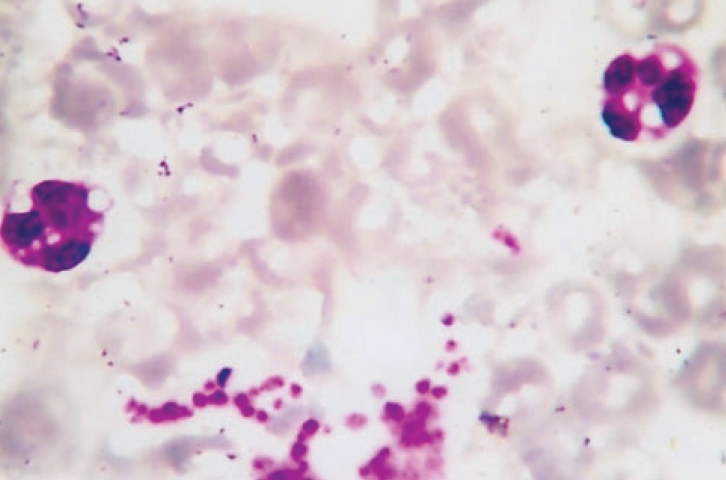 |
| Peripheral blood smear showing lipid vacuoles in leucocytes (Jordans� anomaly) (Wright�s stain, x1000) |
|
| Peripheral blood smear showing lipid vacuoles in leucocytes (Jordans� anomaly) (Wright�s stain, x1000) |
 |
| Thick adherent scales present on the face and trunk |
|
| Thick adherent scales present on the face and trunk |
Introduction
Dorfman-Chanarin syndrome is a multisystem inherited metabolic disorder associated with congenital ichthyosis and accumulation of lipid droplets in various types of cells. The syndrome is also known as ′neutral lipid storage disease with ichthyosis′ because of the deposition of neutral lipids in multiple organs including the skin, muscle, liver, central nervous system and granulocytes.[1] However, there is great variability in dermatologic severity and pattern and degree of systemic involvement. The disease is rare and since the initial case report by Dorfman in 1974,[2] only 30 cases have been reported and majority of them are from the Middle East. We report a case of Dorfman-Chanarin syndrome in a four-year-old girl who had generalized ichthyosis since birth and abdominal distension and episodic vomiting since two months of age. Neutrophils, eosinophils and monocytes in peripheral blood smear revealed lipid vacuoles. Prior to this, there have been three case reports from India.[3],[4],[5]
Case report
A four-year-old girl was brought to the dermatology outpatient department for generalized scaling and erythema present since birth. The scaling had started over the trunk and had subsequently involved the entire body including the scalp and the face within one week. No bullous lesions or erosions had appeared at any point of time. The parents had also noticed abdominal distension in the child. She had developed episodic vomiting at two months of age that gradually became less frequent over a period of 1 year.
However, there was no history of difficulty in feeding, icterus, dark-colored urine or clay-colored stools. No history of ocular complaints, hearing impairment or gait abnormality was reported. She was the second child and was born at full term by normal delivery to non-consanguineous parents. There was no history of prolonged or obstructed labor or history of spontaneous abortion or stillbirth in the mother. There was no history of maternal infection or exposure to drugs, radiation, smoking or alcohol by the mother during pregnancy. No other family member had similar illness.
General physical examination was within normal limits. Cutaneous examination revealed presence of generalized scaling with mild background erythema all over the body including the face and the scalp [Figure - 1]. There was marked involvement of flexures in the form of corrugated appearance, hyperpigmentation and scaling. Individual scale over the trunk was grayish white, fine, translucent and semi-adherent while that on the limbs was dark brown to black, larger in size, polygonal and adherent. The eyelids, lips and ears were involved showing mild ectropion and mild eclabion [Figure - 1]. There was no evidence of keratosis pilaris. The palms, soles, hair, teeth and nails appeared normal. A portwine stain of size 8x10 cm was present over the left side of the chest wall. There was mild xerosis in the left eye but the rest of the ocular examination, including slit-lamp and fundoscopy did not reveal any abnormality. There was no evidence of vertigo or nystagmus to suggest the involvement of the vestibular system. Evaluation of cochlear functions using tuning fork tests and pure tone audiometry was attempted but the child did not co-operate, however, other tests like distraction technique and free-field audiometry did not reveal any abnormality in cochlear functions. The facility for brainstem evoked response audiometry was not available in our institution.
Abdominal examination revealed generalized distension. There was firm, non-tender hepatomegaly and the liver was palpable 3 cm below the costal margin in the mid-clavicular line. Spleen was not palpable. There was no free fluid and the bowel sounds were normal. There was no neurological deficit and muscle tone, power and deep tendon reflexes were normal. There was no gait abnormality nor was there any proximal muscle weakness or tenderness. Rest of the systemic examination was within normal limits.
A complete hemogram, renal function tests, blood sugar and electrolytes were within normal limits. The coagulation profile was normal. However, serum transaminase levels were raised, SGOT being 73 IU/liter (normal 4-40 IU/liter). Serum levels of alkaline phosphatase (856KAU, normal 20-400KAU) and creatine phosphokinase (NAC, 856U, normal 20-200U/L) were found to be elevated. A peripheral blood smear of the patient showed lipid vacuoles in neutrophils, eosinophils and monocytes [Figure - 2]. Abdominal sonography revealed moderate hepatomegaly with increased echotexture suggestive of fatty liver.
Skin biopsy showed acanthosis, hyperkeratosis and lipid vacuoles in the basal layer. A needle biopsy from the liver showed inflammatory infiltrate and moderate fibrosis in portal tracts and diffuse severe fatty change in hepatocytes. Peripheral blood smear examination of both parents and siblings to look for lipid vacuoles in neutrophils and eosinophils was found to be negative. The patient has been put on a generally low-fat diet with minimum amount of saturated fat in consultation with a dietician. She was started on emollients to which she responded only minimally. Systemic administration of acitretin was contemplated; however, it could not be started in view of the deranged liver function tests.
Discussion
Dorfman-Chanarin syndrome is characterized by the presence of congenital ichthyosis with deposition of lipid droplets in multiple organs. There is an increased fibroblast triglyceride synthesis with failure of triglyceride breakdown[6] leading to the deposition of neutral lipids in various cells and organs viz. skin, muscles, liver, central nervous system, gastrointestinal tract (rectal, gastric and small bowel mucosa), granulocytes, monocytes, mast cells, megakaryocytes, pericytes, muscle cells, Schwann cells and in the bone marrow in promyelocytes, myelocytes and metamyelocytes.[1],[2],[4],[6],[7],[8] This may lead to myriad clinical features including neurological abnormalities, myopathy, growth retardation, sensorineural deafness, cataract, strabismus and retinal dysfunction besides congenital ichthyosis.[1],[4] Various neurological abnormalities associated with the Dorfman-Chanarin syndrome include nystagmus, ataxia, areflexia, diffuse hypotonia, marked proximal muscle weakness, myopathic gait, ptosis, cranial nerve weakness, mental retardation and psychiatric disorders.[2],[9] Splenomegaly, neural tumor of the mediastinum and microcephaly were reported to be associated with this syndrome by Srebrnik et al.[6],[9]
The first case of Dorfman-Chanarin syndrome was described by Dorfman in 1974.[2] Prior to that, Jordans[10] had reported two brothers with Erb′s type of progressive muscular dystrophy whose peripheral blood leucocytes contained fat vacuoles (Jordans′ anomaly). Rozenszajn[7] and coworkers described a similar Jordans′ anomaly in 1966 in two sisters who also suffered from ichthyosis. Lefevre identified CGI-58 mutations in nine families, all of them from the Mediterranean region.[11] CGI-58 is a member of the esterase/lipase/thioesterase subfamily of proteins and is involved in the lipid metabolism of lamellar granules. A seriously truncated CGI-58 protein leads to the defective formation of lamellar granules in the human epidermis. Later, this CGI-58 mutational defect in a Dorfman-Chanarin syndrome patient was also reported from the nonMediterranean region by Akiyama in 2003.[12] A mutation in the ABHD5 (Abhydrolase domain containing 5) gene has been identified to be the genetic basis for this disease, although the precise function of the enzyme encoded by the gene remains unclear.[13]
Our patient had congenital ichthyosis and Jordans′ anomaly. Enlargement of liver and elevation of liver enzymes indicate presence of fatty change which was later confirmed by ultrasonography and liver biopsy. No muscle weakness or mental retardation was observed in the patient and the milestones till now have been within normal limits. Jordans′ anamoly was seen in the granulocytes and monocytes of our patient. Various previous workers have emphasized the importance of peripheral blood smear examination of clinically unaffected members for Jordans′ anamoly to help in the detection of heterozygous carriers.[3] Conductor analysis on both parents and sibling by doing a peripheral smear was negative in our case. We also screened peripheral blood eosinophils for lipid vacuoles as suggested by Wollenberg et al[8] as an alternative to traditional screening of neutrophils and found the same results.
Response of gastrointestinal symptoms and muscle weakness to a gluten-free diet has been reported.[14] Also, a low-fat diet poor in long-chain fatty acids and rich in medium-chain fatty acids has been reported to improve symptoms.[15] We suggested these dietary modifications to the mother. In view of the young age of child the parents were not keen on incorporating dietary modifications. However, in consultation with the dietician, they have agreed to a low-fat diet with minimum amount of saturated fat.
In addition, ichthyosis in the patient presents a management problem as retinoids (acitretin) which are the systemic therapeutic modality of choice in lamellar ichthyosis cannot be given to this patient in view of her hepatomegaly and deranged liver functions. The unusual features in our case are that the liver function test derangement and hepatomegaly have been non-progressive over the last 4 years during which the patient has been on regular follow-up and periodic investigations. Moreover, there is absence of clinical muscle weakness and there is no evidence of myopathy on muscle enzyme estimation. There are no ocular abnormalities and no neurological deficits on detailed neurological examination. This is unusual as muscle weakness and neurological deficits are commonly associated with the Dorfman-Chanarin syndrome. We have kept the patient on regular follow-up in order to monitor hepatic disease and to rule out late onset of neurological and muscle weakness which may occur at a later stage.
| 1. |
Judge MR, McLean WH, Munro CS. Disorders of keratinization. In : Burns T, Breathnach S, Cox N, Griffiths C, editors. Rook's Textbook of Dermatology. 7th ed. Oxford, London: Blackwell Science; 2004. p. 34.1-34.111.
[Google Scholar]
|
| 2. |
Dorfman ML, Hershko C, Eisenberg S, Sagher F. Ichthyosiform dermatosis with systemic lipidosis. Arch Dermatol 1974;110:261-6.
[Google Scholar]
|
| 3. |
Nanda A, Sharma R, Kanwar AJ, Kaur S, Dash S. Dorfman-Chanarin syndrome. Int J Dermatol 1990;29:349-51.
[Google Scholar]
|
| 4. |
Tullu MS, Muranjan MN, Save SU, Deshmukh CT, Khubchandani SR, Bharucha BA. Dorfman-Chanarin syndrome: A rare neutral lipid storage disease. Indian Pediatr 2000;37:88-93.
[Google Scholar]
|
| 5. |
Gupta P, Kaur G. Chanarin Dorfman syndrome neonatal diagnosis and 3-year follow-up. Indian Pediatr 2005;42:1054-5.
[Google Scholar]
|
| 6. |
Judge MR, Atherton DJ, Salvayre R, Hilaire N, Levade T, Johnston DI, et al . Neutral lipid storage disease: Case report and lipid studies. Br J Dermatol 1994;130:507-10.
[Google Scholar]
|
| 7. |
Rozenszajn L, Klajman A, Yaffe D, Efrati P. Jordans' anomaly in white blood cells. Blood 1966;28:258-65.
[Google Scholar]
|
| 8. |
Wollenberg A, Geiger E, Schaller M, Wolff H. Dorfman-Chanarin syndrome in a Turkish kindred: Conductor diagnosis requires analysis of multiple eosinophils. Acta Derm Venereol 2000;80:39-43.
[Google Scholar]
|
| 9. |
Srebrnik A, Tur E, Perluk C, Elman M, Messer G, Ilie B, et al . Dorfman-Chanarin syndrome: A case report and a review. J Am Acad Dermatol 1987;17:801-8.
[Google Scholar]
|
| 10. |
Jordans GH. The familial occurrence of fat-containing vacuoles in the leucocytes diagnosed in two brothers suffering from dystrophia musculorum progressiva. Acta Med Scand 1953;145:419-23.
[Google Scholar]
|
| 11. |
Lefevre C, Jobard F, Caux F, Bouadjar B, Karaduman A, Heilig R, et al . Mutations in CGI-58, the gene encoding a new protein of the esterase/lipase/thioesterase subfamily, in Chanarin-Dorfman syndrome. Am J Hum Genet 2001;69:1002-12.
[Google Scholar]
|
| 12. |
Akiyama M, Sawamura D, Nomura Y, Sugawara M, Shimizu H. Truncation of CGI-58 protein causes malformation of lamellar granules resulting in ichthyosis in Dorfman-Chanarin Syndrome. J Invest Dermatol 2003;121:1029-34.
[Google Scholar]
|
| 13. |
Doganci T, Gurakar F, Karaduman A, Orhan D, Caglar M. Images of interest. Hepatobiliary and pancreatic: Dorfman-Chanarin syndrome. J Gastroenterol Hepatol 2005;20:156.
[Google Scholar]
|
| 14. |
Miranda A, DiMauro S, Eastwood A, Hays A, Johnson WG, Olarte M, et al . Lipid storage myopathy, ichthyosis and steatorrhea. Muscle Nerve 1979;2:1-13.
[Google Scholar]
|
| 15. |
Kakourou T, Drogari E, Christomanou H, Giannoulia A, Dacou-Voutetakis C. Neutral lipid storage disease - response to dietary intervention. Arch Dis Child 1997;77:184.
[Google Scholar]
|
Fulltext Views
5,629
PDF downloads
4,393





![[Figure - 1]](#fig_ijdvl_2007_73_1_36_30650_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2007_73_1_36_30650_2.jpg){kind=link}