
Ectodermal dysplasia-skin fragility syndrome – identification of a novel plakophilin1 (PKP1) gene variant through whole exome sequencing
 , Amritha Konda3,, Vadlamudi Raghavendra Rao4, Vilas Adepu5
, Amritha Konda3,, Vadlamudi Raghavendra Rao4, Vilas Adepu5
Corresponding author: Dr. Sharath Chandra Konda, Department of Dermatology, SVS Medical College, Yenugonda, Telangana, India. drsharath2k@yahoo.co.in
-
Received: ,
Accepted: ,
How to cite this article: Konda SC, Biswas A, Konda A, Rao VR, Adepu V. Ectodermal dysplasia-skin fragility syndrome – identification of a novel plakophilin1 (PKP1) gene variant through whole exome sequencing. Indian J Dermatol Venereol Leprol. 2025;91:381-5. doi: 10.25259/IJDVL_420_2023
Dear Editor,
A 16-year-old boy, presented with generalised skin erosions, dryness and cracking of lips, thickened skin on the palms and soles, abnormal nails, and woolly hair. The symptoms started at the age of 3 months with gradual progression. He was the first child of a second-degree consanguineous marriage, with his younger sister and both parents being asymptomatic. On physical examination, his height (147 cm) and weight (33 kg) were below the third centile for his age, with a normal gait. Cutaneous examination revealed generalised xerosis with features of skin fragility in the form of numerous superficial skin erosions with crusting on the upper back [Figure 1a], chest, and limbs, with the face being relatively spared. Palmoplantar keratoderma was present with superficial fissures [Figures 1b and 1c]. The scalp showed coarse woolly hair, without alopecia/hypotrichosis, but with remarkably easy pluckability [Figure 1d]. The eyebrows looked thick and dense, with the eyelashes being normal. The lips and perioral region were red, scaly, and fissured [Figure 1e]. Nails were dystrophic with subungual hyperkeratosis and distal curving. A general examination of all other systems, including the cardiovascular system, was normal.

- Superficial erosions on the upper trunk.
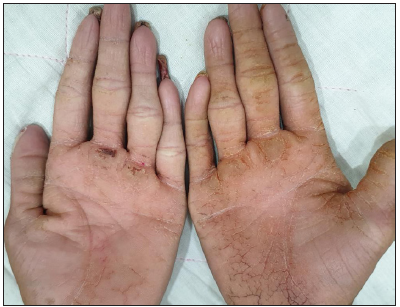
- Palmar keratoderma.
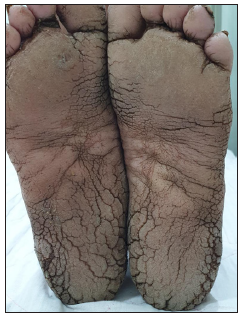
- Plantar keratoderma.
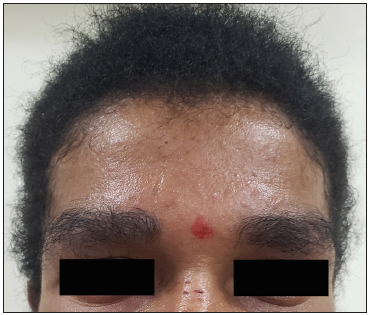
- Scalp and eyebrows showing woolly hair.
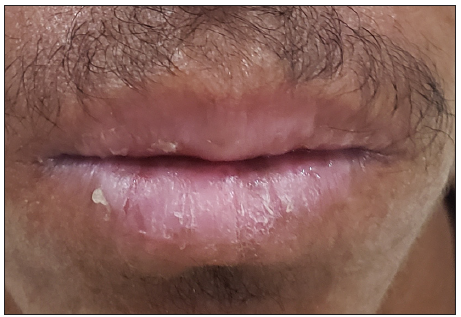
- Erosions and scaling of lips.
A trichogram of plucked hair showed dystrophic anagen hair. Histopathology revealed widened intercellular spaces with a split in the sub-corneal layer [Figure 2a].
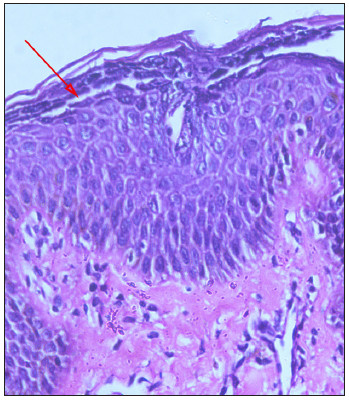
- Histopathology of skin ((Haematoxylin and Eeosin 40x) staining under 40 magnification) with arrow showing the blister.
Scanning electron microscopy (SEM) of hair revealed damaged, rough cuticles with paint brush fractures of the cortex and dystrophic anagen roots [Figures 2b and 2c].
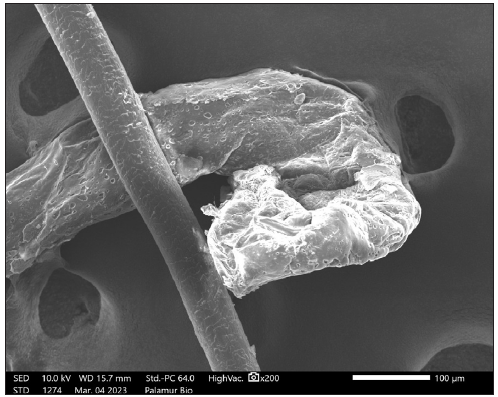
- Scanning electron microscopic (SEM) image of hair showing dystrophic anagen root.

- Scanning electron microscopic image of hair showing fractured cortex.
Whole-exome sequencing identified a novel missense variant, c.1061T>C (p. Leu354Pro) in exon 6 of plakophilin 1 gene. This variant was validated using sanger sequencing in the proband, parents, and younger sister. Both the parents and the sister were heterozygous, whereas the proband was homozygous for the mutant variant [Figures 3a and 3b]. Pathogenicity prediction software tools predicted this variant to be deleterious. The 3D structure of the wild and mutant protein showed a change in the orientation of amino acid–producing destabilizing effects [Figure 3c].

- Family pedigree: Gray box shows the affected proband.
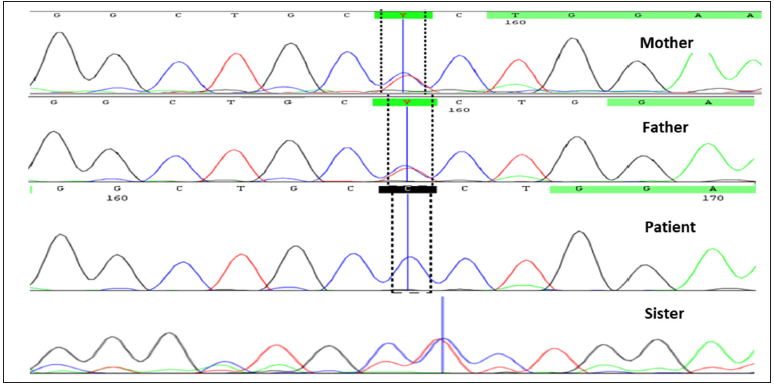
- Sanger sequencing showing homozygous variation in the proband as compared to parents and sibling.
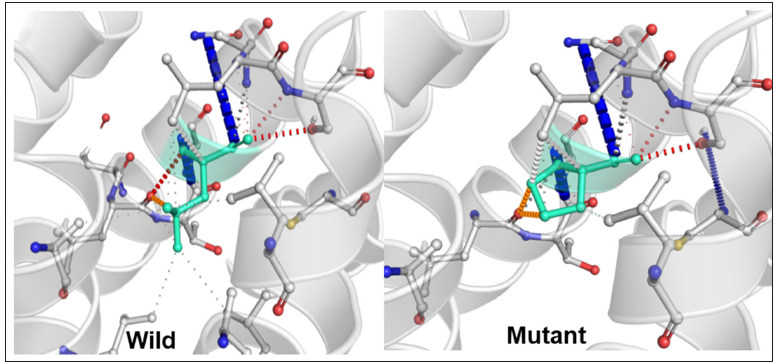
- Destabilizing change in orientation of protein structure due to the presence of a mutation.
The differential diagnoses considered were generalised epidermolysis bullosa simplex (excluded by lack of extensive blisters), Naxos and Carvajal syndrome (excluded by lack of cardiomyopathy), and Kindler epidermolysis bullosa (excluded by lack of photosensitivity and poikiloderma).
As there are no specific treatments for this condition, the boy was managed conservatively with moisturisers and topical antibacterials for skin lesions, lip balms for the lips, and keratolytic creams for the palms and soles.
Discussion
Desmosomes are junctions essential for preserving tissue integrity by tethering adjoining cells and anchoring the cell membrane to the keratin intermediate filament cytoskeleton internally. These are intercellular junctional complexes primarily found in epithelial tissues but also in meninges, lymph node follicles, and myocardium.1 The main structural constituents of desmosomes are a) the cadherins – desmogleins and desmocollins; b) the plakins – desmoplakin, periplakin, and envoplakin; and c) the armadillo family of proteins – plakoglobin, plakophilins (PKP1 and 2). These interact with one another near the plasma membrane and with keratin intermediate filaments in the cytoplasm. Ectodermal dysplasia-skin fragility syndrome, first described by McGrath et al.2 is a rare autosomal recessive genodermatosis characterised by cutaneous erosions/blisters, palmoplantar keratoderma, hair abnormalities/alopecia, characteristic lip scaling with fissures and nail dystrophy resulting from loss of function mutations in the Plakophilin 1 (PKP1) gene.2
An ectodermal dysplasia-skin fragility syndrome patient born out of consanguineous marriage and possessing a homozygous variant in the PKP1 gene causing a substitution of leucine with proline at codon 1061 is described here. In the literature review of all reported cases [ Table 1], homozygosity was present in 87% and parental consanguinity was present in 66% of cases, including the present case. Twenty three cases have been reported with confirmed bi-allelic mutations in the PKP1 gene, ours being the latest. Structurally, PKP1 is located close to the plasma membrane in the desmosomal plaque, binding and interacting primarily with desmoplakin (via the amino-terminal domain).2,3
| S. No | Age (years)/Gender | Origin/Ethnicity | Parental consanguinity | Zygosity | Variant, AA change | Variation |
|---|---|---|---|---|---|---|
| 12 | 6/M | British | No | Compound heterozygous |
c.910C>T – p.Gln304* c.1132ins28 – NA |
Nonsense Frameshift |
| 23 | 0.5/F | Arab | Yes | Homozygous | c.847-2A>G – N/A | Splice site |
| 33 | 3/F | Arab | Yes | Homozygous | c.203-1G>A – N/A | Splice site |
| 45 | 1/F | Egyptian (case 1) | Yes | Homozygous | c409_410insAC – p.Thr137Thrfs*61 | Frameshift |
| 55 | 0.9/M | Egyptian (case 2) | No | Homozygous | c1213delA – p.Arg411Glufs*22 | Frameshift |
| 66 | 8/M | Chinese | No | Homozygous | c.723delG –p.E241Dfs*4 | Frameshift |
| 77 | 1.5/M | British | No | Compound heterozygous | c.203-1G>A – N/A c.213T>G - p.Tyr71* |
Splice site Nonsense |
| 88 | 17/M | British | No | Homozygous | c.1233-2A>T – N/A | Splice site |
| 99 | 42/M | Japan | Yes | Homozygous | c.2021+1G>A – N/A | Splice site |
| 1010 | 33/M | Dutch | N/A | Homozygous | c1680+1G>A – N/A | Splice site |
| 1111 | 3/F | Chinese | No | Compound heterozygous |
c.1835-2A>G – N/A c.1053T>A+1054 – N/A +1G>T |
Splice site |
| 1212 | 6/M | Turkish | Yes | Homozygous | c.888delC-p.Arg297Alafs*42 | Frameshift |
| 1313 | 10/M | Brazilian | Yes | Homozygous | c.2014C>T – p.Arg672* | Nonsense |
| 1414 | 1.2/F | Iraqi | Yes | Homozygous | c.897del5 – p.Asn300Glufs*60 | Frameshift |
| 1515 | 15/M | Spanish | N/A | Homozygous | c.1233-2A>G – N/A | Splice site |
| 1616 | 5/F | Egyptian | Yes | Homozygous | c.203-1G>T – N/A | Splice site |
| 1716 | 4/F | Egyptian | Yes | Homozygous | c.203-1G>T – N/A | Splice site |
| 1817 | 27/F | Turkish | Yes | Homozygous | c.1411-9G>A – N/A | Splice site |
| 1918 | 29/M | Turkish | Yes | Homozygous | c.1414_1415delTG – p.Val472Glyfs*28 | Frameshift |
| 2019 | 2/F | Caucasian | No | Homozygous in lesional skin | c.638delT – p.V213Gfs*33 | Frameshift |
| 2120 | 5/M | Chinese | N/A | Homozygous | c.203-1G>A – N/A | Splice site |
| 2221 | 16/F | Spanish | Yes | Homozygous | c.455C>T – p.Ala152Val | Missense |
| 2321 | Neonate/F | Spanish | Yes | Homozygous | c.455C>T – p.Ala152Val | Missense |
| 24** | 16/M | Indian | Yes | Homozygous | C1061T>C p.Leu354Pro | Missense |
PKP 1: Plakophilin 1, EDSF: Ectodermal dysplasia skin fragility syndrome, M: Male, F: Female, N/A: Not available, AA: Amino acid, *: stop codon, **: present study.
Desmosomal PKP1 distribution is concentrated in the suprabasal layers of stratified and complex epithelia such as skin, outer root sheath cells of hair follicles, etc. 4 PKP1 is a multifunctional protein promoting desmosomal stability. 5 In previously reported studies, skin fragility, and nail involvement were present in all 23 cases, as in our case. Perioral and lip erythema, scaling and fissures were reported in 18/23 cases, palmoplantar keratoderma in 21/23 cases and alopecia/hypotrichosis/woolly hair in 22/23 cases. Additionally, hypohidrosis was reported in 6/23 cases and pruritus, dental anomalies, recurrent cutaneous infections, pneumonia, delayed milestones, and low height and weight centiles were present in a few cases. 3 We observed that ectodermal dysplasia-skin fragility syndrome occurred in all ethnic populations [ Table 1 ]. Mutations in the head domain (5’N-terminal) resulted in absent or markedly decreased PKP1 with an early onset of symptoms that were severe in nature, especially skin fragility (average age 2.9 years). Further down mutations towards the C-terminal/3’end resulted in reduced PKP1 and milder symptoms with later onset) (average age 28.2 years). Mutations in the armadillo repeat units 1, 2, and 3 were associated with woolly/curly/wiry hair, including our case.
Perioral and lip scaling and fissuring is a persistent and prominent distinguishing feature that should prompt the clinician to consider ectoderma dysplasis-skin fragility syndrome. Hair pathology varied from total alopecia to sparse and woolly hair, which is due to abnormal or absent PKP1 in the suprabasal cells of the outer root sheath of hair follicles. The hair in our patient was normal at birth but subsequently became woolly and was strikingly easily pluckable, which was not reported in any other case. Similar trichogram and scanning electron microscopy hair findings were reported in 2020 by Sun et al.6
Conclusion
PKP1 mutations, though rare, constitute an important disorder of desmosomes with core clinical features of skin fragility, lip, and perioral scaling, nail abnormalities, palmoplantar keratoderma and alopecia/hypotrichosis/woolly hair. We herewith report a case of ectodermal dysplasia-skin fragility/McGrath syndrome in an Indian boy with a novel homozygous missense mutation in the PKP1 gene, in exon 6 in the armadillo repeat region motif-3 with a review and expansion of current literature on the PKP1 mutation database and phenotype–genotype correlation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Plakophilin 1: An important stabiliser of desmosomes. Clin Exp Dermatol. 2004;29:161-7.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet. 1997;17:240-4.
- [CrossRef] [PubMed] [Google Scholar]
- Homozygous splice site mutations in PKP1 result in loss of epidermal plakophilin 1 expression and underlie ectodermal dysplasia/skin fragility syndrome in two consanguineous families. J Invest Dermatol. 2004;122:647-51.
- [CrossRef] [PubMed] [Google Scholar]
- Ectodermal dysplasia-skin fragility syndrome. Indian J Dermatol Venereol Leprol. 2011;77:503-6.
- [CrossRef] [PubMed] [Google Scholar]
- Ectodermal dysplasia-skin fragility syndrome: Two new cases and review of this desmosomal genodermatosis. Exp Dermatol. 2020;29:520-30.
- [CrossRef] [PubMed] [Google Scholar]
- Novel homozygous deletion of the plakophilin-1 gene in a Chinese patient with ectodermal dysplasia–skin fragility syndrome. J Dermatol. 2020;47:779-81.
- [CrossRef] [PubMed] [Google Scholar]
- Skin fragility and hypohidrotic ectodermal dysplasia resulting from ablation of plakophilin 1. Br J Dermatol. 1999;140:297-307.
- [CrossRef] [PubMed] [Google Scholar]
- Genomic amplification of the human plakophilin 1 gene and detection of a new mutation in ectodermal dysplasia/skin fragility syndrome. J Invest Dermatol. 2000;115:368-74.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype-phenotype correlation in skin fragility-ectodermal dysplasia syndrome resulting from mutations in plakophilin 1. Exp Dermatol. 2002;11:107-14.
- [CrossRef] [PubMed] [Google Scholar]
- J Invest Dermatol. 2004;122:1321-4.
- [CrossRef] [PubMed]
- Compound heterozygosity for new splice site mutations in the plakophilin 1 gene (PKP 1) in a Chinese case of ectodermal dysplasia-skin fragility syndrome. Acta Derm Venereol. 2005;85:394-9.
- [CrossRef] [PubMed] [Google Scholar]
- Ectodermal dysplasia-skin fragility syndrome resulting from a new homozygous mutation, 888delC, in the desmosomal protein plakophilin 1. J Am Acad Dermatol. 2006;55:157-61.
- [CrossRef] [PubMed] [Google Scholar]
- Novel truncating mutations in PKP 1 and DSP cause similar skin phenotypes in two Brazilian families. Br J Dermatol. 2009;160:692-7.
- [CrossRef] [PubMed] [Google Scholar]
- Ectodermal dysplasia-skin fragility syndrome due to a new homozygous internal deletion in the PKP1 gene. Australas J Dermatol. 2012;53:61-5.
- [CrossRef] [PubMed] [Google Scholar]
- Ectodermal dysplasia-skin fragility syndrome: A novel mutation in the PKP1 gene. Clin Exp Dermatol. 2013;38:787-90.
- [CrossRef] [PubMed] [Google Scholar]
- A plakophilin-1 gene mutation in an Egyptian family with ectodermal dysplasia-skin fragility syndrome. Mol Syndromol. 2014;5:304-6.
- [CrossRef] [PubMed] [Google Scholar]
- J Dermatol Sci. 2016;84:210.
- [CrossRef] [PubMed]
- Ectodermal dysplasia-skin fragility syndrome with a new mutation. Indian J Dermatol Venereol Leprol. 2017;83:476-9.
- [CrossRef] [PubMed] [Google Scholar]
- A case of mosaicism in ectodermal dysplasia-skin fragility syndrome. Br J Dermatol. 2017;177:101-2.
- [Google Scholar]
- Histopathological and genetical diagnosis of one case of neonatal ectodermal dysplasia/skin fragility syndrome. Zhonghua Shao Shang Za Zhi. 2020;36:500-2.
- [CrossRef] [PubMed] [Google Scholar]
- Ectodermal dysplasia-skin fragility syndrome: Two new cases with a novel missense mutation. J Dtsch Dermatol Ges. 2020;19:595-7.
- [CrossRef] [PubMed] [Google Scholar]




