Translate this page into:
Epidermolysis bullosa acquisita
Correspondence Address:
Jayanta Kr Das
Department of Dermatology, Ramakrishna Mission Seva Pratisthan, 99, Sarat Bose Road, Kolkata 700026, West Bengal
India
| How to cite this article: Das JK, Sengupta S, Gangopadhyay AK. Epidermolysis bullosa acquisita. Indian J Dermatol Venereol Leprol 2006;72:86 |
 |
|
 |
|
 |
|
Epidermolysis bullosa (EB) generally refers to a group of inherited disorders that involve the formation of blisters following trivial trauma.[1] EB acquisita (EBA) is a chronic subepidermal blistering disease associated with autoimmunity to type-VII collagen within anchoring fibrils located at the dermal-epidermal junction.[2],[3] Although an acquired disease, it was placed in the category of EB approx 100 years ago as physicians were struck by its similarity with hereditary dystrophic EB.[2],[3] This is an entity rarely reported from India.[3] Here is the report of a case with pruritus, an atypical feature.
CASE REPORT
A 41-year-old housewife presented with pruritic blisters on both lower limbs, and a few on the elbows. The blisters used to appear since the age of 26 years following mild trauma, and healed with scars. They had first appeared on the knees and shins, and later on the elbows and dorsal aspect of the hands, and rarely on the buttocks. Subsequently, lesions on the shins, and a few on the knees and elbows persisted, whereas those on other areas had subsided. They were pruritic to start with.
The patient was born of a non-consanguineous marriage. She had no such lesions in her childhood. None of her family members, including her four sisters, three brothers, and five children, had similar lesions. There was no history suggestive of chronic systemic illness, including inflammatory bowel disease, diabetes, collagen vascular diseases, or other autoimmune diseases.
On examination, many small, tense blisters and a few scars were seen on the shins, dorsal aspect of the feet, above the medial malleolus, knees [Figure - 1] and elbows. There was a tense hemorrhagic blister at the base of the left third toe. There were no signs of inflammation or prurigo-like lesions. There were no milia or albopapuloid lesions. Both great toenails were dystrophic [Figure - 2]. Her mucosae, hair, scalp, palms, and soles were free from any lesions, and the teeth were normal. Her systemic examination was unremarkable.
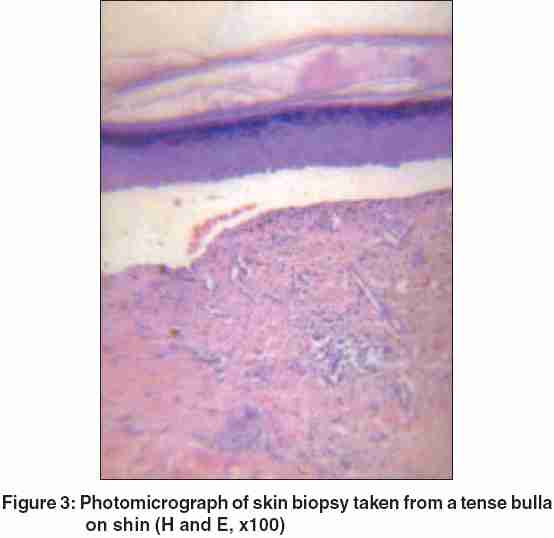
Routine blood, urine, and stool examinations, liver function tests, and urine porphyrins were within normal limits. No fungus was found in nail clippings of the clinically dystrophic nails. Histopathological examination of lesional skin showed a subepidermal blister with clear-cut separation between the dermis and epidermis, and a minimal inflammatory infiltrate [Figure - 3]. Direct immunofluorescence testing of perilesional skin showed a linear fluorescent band of immunoglobulin G (IgG) at the dermal-epidermal junction. Electron microscopy could not be done because this facility was not available.
The patient was made to understand the importance of avoiding trauma. The blisters and ulcers were dressed regularly, and oral antibiotics were given whenever required. She was first put on colchicine, but as she had nausea and diarrhea at doses above 0.6 mg/day and was without any response, methotrexate was started. Oral courses of methotrexate, started at a dose of 15 mg/week, and then increased to 30 mg/week, were not helpful, and the patient was neither regular in follow-up nor willing to undergo repeated tests that were necessary for treatment with methotrexate. Hence, she was treated mainly with supportive therapy, which she felt was giving her acceptably good relief. After about 6 months of treatment she was lost to follow-up.
DISCUSSION
The localization of immune deposits within the dermal-epidermal junction of the skin of EBA patients by immunoelectron microscopy is considered the gold standard for the diagnosis.[2] Ultrastructural studies of EBA show a reduction of anchoring fibrils and an amorphous, electron-dense band just beneath the lamina densa. As we could not perform electron microscopy to confirm the diagnosis of EBA, the widely accepted diagnostic criteria proposed by Yaoita et al .[2] could not be applied. Instead, our patient was diagnosed as having EBA as it satisfied the criteria of Roenigk et al.:[3] (a) a negative family and personal history of previous blistering disease, (b) an adult-onset eruption, (c) spontaneous or trauma-induced blisters that resemble those of hereditary dystrophic EB and (d) exclusion of all other bullous diseases. Our patient had non-inflammatory blisters following trauma, at sites prone to trauma, repeatedly for 15 years since the age of 26 years. Nail dystrophy was present. The histopathology and direct immunofluorescence findings were consistent with EBA. The clinical features were not suggestive of any other bullous disease. Thus, this clearly was a case of EBA.
Clinically, EBA may mimic a number of bullous dermatoses. Blisters may be serous or hemorrhagic, tend to be localized to areas of trauma (especially the dorsal aspect of the hands and feet, and elbows), and heal with scarring, milia formation, and hyperpigmentation. The nails may be dystrophic but are often normal. Mucosal involvement is variable.[4]
Pruritus was a predominant feature in our patient. Although pruritus has not been mentioned as a typical manifestation of EBA, it is the recognized feature of some of the types of inherited EB.[5],[6] Microscopic studies of EB pruriginosa, a rare form of inherited EB, show typical findings of dystrophic EB,[7] and it has been postulated that itching lesions of EB pruriginosa could represent an abnormal dermal reactivity of some subjects to their inherited bullous disorder. Itching in EBA may also be owing to the same reason.
EBA is a very difficult disease to treat. Supportive therapy, such as instructions in wound care and strategies for avoiding all sorts of trauma, is warranted in all patients of EBA. The patients must be educated to recognize skin infection and to seek medical care and antibiotic therapy promptly. Systemic glucocorticoids, azathioprine, methotrexate, and cyclophosphamide are mostly ineffective, even in large doses. Some patients improve with dapsone, especially when neutrophils are present in their dermal infiltrate. Cyclosporine is often effective, but has long-term toxicity. Colchicine in high doses is helpful in many cases, provided the patient can tolerate its gastrointestinal side effects.[2]
Although electron microscopy is needed for confirming the diagnosis of EBA, it could not be performed in any of the cases reported earlier from India.[3],[8],[9] We report on this case as it satisfies the clinical and histological criteria for the diagnosis of this rare disease, and shows pruritus as an atypical feature.
| 1. |
Marinkovich MP, Khavari PA, Herron GS, Bauer EA. Inherited epidermolysis bullosa. In : Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Dermatology in General Medicine. 6th ed. New York: McGraw-Hill; 2003. p. 596-609.
[Google Scholar]
|
| 2. |
Woodley DT, Chen M, Gammon WR, Briggaman RA. Epidermolysis bullosa acquisita. In : Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Dermatology in General Medicine. 6th ed. New York: McGraw-Hill; 2003. p. 609-16.
[Google Scholar]
|
| 3. |
Gangopadhyay AK. Epidermolysis bullosa acquisita. Indian J Dermatol 1997;42:121-2.
[Google Scholar]
|
| 4. |
Wojnarowska F, Eady RA, Burge SM. Bullous eruptions. In : Champion RDH, Burton JL, Burn DA, Breathnach SM, editors. Rook/Wilkinson/Ebling's Textbook of Dermatology. 6th ed. Oxford: Blackwell Science; 1998. p. 1817-97.
[Google Scholar]
|
| 5. |
McGrath JA, Schofield OM, Eady RA. Epidermolysis bullosa pruriginosa: dystrophic epidermolysis bullosa with distinctive clinicopathological features. Br J Dermatol 1994;130:617-25.
[Google Scholar]
|
| 6. |
Goulden V, Handfield-Jones S, Neild V, Black MM. Linear prurigo simulating dermatitis artefacta in dominant dystrophic epidermolysis bullosa. Br J Dermatol 1993;129:443-6.
[Google Scholar]
|
| 7. |
Cambiaghi S, Brusasco A, Restano L, Cavalli R, Tadini G. Epidermolysis bullosa pruriginosa. Dermatology 1997;195:65-8.
[Google Scholar]
|
| 8. |
Raj R, Srinivas CR, Mustafa M, Surendran P. Epidermolysis bullosa acquisita in a young female. Indian J Dermatol Venereol Leprol 1999;65:296-7.
[Google Scholar]
|
| 9. |
Fernandes CZ, Bhat MR. Epidermolysis bullosa acquisita-a case report. Indian J Dermatol 2002;47:96-7.
[Google Scholar]
|
Fulltext Views
4,293
PDF downloads
1,969





![[Figure - 1]](#fig_ijdvl_2006_72_1_86_19736_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2006_72_1_86_19736_2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2006_72_1_86_19736_3.jpg){kind=link}