Translate this page into:
Epidermolysis bullosa acquisita and anti-p200 pemphigoid as major subepidermal autoimmune bullous diseases diagnosed by floor binding on indirect immunofluorescence microscopy using human salt-split skin
2 Department of Dermatology, Kasturba Medical College, Manipal University, Mangalore, Karnataka, India
3 Department of Immunodermatology, St. John's Institute of Dermatology, St Thomas' Hospital, London, UK
4 Department of Dermatology, University of Lübeck, Lübeck, Germany
Correspondence Address:
Raghavendra Rao
Department of Dermatology, Kasturba Medical College, Manipal University, Manipal - 576 104, Karnataka
India
| How to cite this article: Goyal N, Rao R, Shenoi SD, Pai S, Kumar P, Bhogal BS, Schmidt E, Zillikens D. Epidermolysis bullosa acquisita and anti-p200 pemphigoid as major subepidermal autoimmune bullous diseases diagnosed by floor binding on indirect immunofluorescence microscopy using human salt-split skin. Indian J Dermatol Venereol Leprol 2017;83:550-555 |
Abstract
Background: Subepidermal autoimmune bullous diseases are a diverse group of diseases with overlapping clinical and immunopathological features. Indirect immunofluorescence microscopy on artificially split skin helps to classify these conditions into those with staining on the epidermal side of the split (“roof-binding”) and those with staining on the dermal side (“floor-binding”). Epidermolysis bullosa acquisita is the prototype of “floor-binding” subepidermal autoimmune bullous diseases. However, not all floor-binding sera are associated with epidermolysis bullosa acquisita.Aim: The aim of this study was to evaluate the clinical and immunological profile of patients with floor-binding subepidermal autoimmune bullous disease by indirect immunofluorescence microscopy and to identify the target antigens in them.
Methods: Ten patients who showed a floor-binding pattern were studied with regard to their clinical and immunopathological characteristics. Target antigens were identified by modified indirect immunofluorescence microscopy using recessive dystrophic epidermolysis bullosa skin, enzyme linked immunosorbent assay, and immunoblotting.
Results: Diagnosis of epidermolysis bullosa acquisita was confirmed in six patients. Three patients with an inflammatory subepidermal autoimmune bullous disease mimicking bullous pemphigoid reacted with a 200 kDa protein on immunoblotting with dermal extract, as is characteristic of anti-p200 pemphigoid. One serum showed both roof and floor binding, and reacted with the BP180 antigen.
Limitation: We could not perform serration pattern analysis in our patients.
Conclusion: In this study, we report three cases of anti-p200 pemphigoid from India. These cases, though indistinguishable clinically from bullous pemphigoid, revealed a floor-binding pattern on indirect immunofluorescence using salt-split skin.
Introduction
The subepidermal autoimmune bullous diseases are a heterogeneous group of conditions characterized by antibody formation against structural proteins of the dermal-epidermal junction resulting in separation of the epidermis from the dermis. They present clinically with tense blisters and erosions on the skin with or without mucous membrane involvement.[1] Lesional histopathology shows subepidermal splitting with a granulocyte-rich inflammatory infiltrate in the upper dermis. Direct immunofluorescence microscopy of a perilesional skin biopsy characteristically shows linear staining of the basement membrane zone with immunoglobulin G, C3, and/or immunoglobulin A in all the subepidermal autoimmune bullous diseases.[2] Indirect immunofluorescence microscopy on salt-split skin further helps categorize these conditions into either “roof”- or “floor”-binding diseases. Bullous pemphigoid is the prototype roof-binding disease while epidermolysis bullosa acquisita typically shows a floor-binding pattern.[3] However, not all patients with floor-binding subepidermal autoimmune bullous disease have epidermolysis bullosa acquisita; anti-laminin 332 mucous membrane pemphigoid which accounts for about 20% of mucous membrane pemphigoid patients, and anti-p200 pemphigoid also exhibit staining on the dermal side of artificially split skin.[4],[5],[6],[7],[8],[9]
This study was undertaken to evaluate the clinical and immunological profile of patients with floor-binding subepidermal autoimmune bullous disease seen at one centre, and to identify the target antigen in this subgroup of patients by indirect immunofluorescence microscopy using recessive dystrophic epidermolysis bullosa skin, enzyme linked immunosorbent assay, and immunoblotting.
Methods
The study was approved by the Institutional Ethical Committee Review Board. Patient consent was obtained in each case.
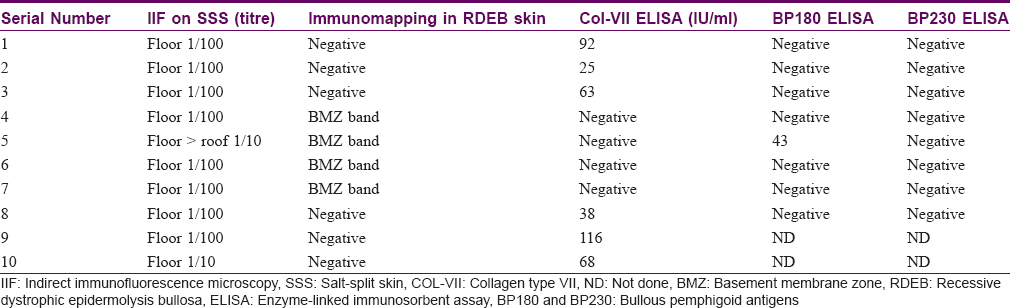
Sera of ten Indian patients received in the immunofluorescence laboratory of Kasturba Medical College, Manipal, between July 2012 and July 2014 were included in the study. All patients showed a floor-binding pattern on indirect immunofluorescence microscopy with salt-split skin. Information pertaining to demographics, clinical profile, treatment and follow-up was taken from hospital records and noted in a predesigned pro forma.
A multistep protocol was followed to identify the target antigens. Initially, modified indirect immunofluorescence microscopy was carried out using frozen sections of skin deficient in type VII collagen obtained from a patient with severe recessive dystrophic epidermolysis bullosa, a technique described previously.[10] Lack of type VII collagen expression was verified by negative staining with monoclonal antibody LH7.2 (Sigma, St. Louis, MO, USA). The frozen sections were then incubated with the patient's serum in 1:10 dilution for 1 h, followed by routine indirect immunofluorescence microscopy.
Subsequently, sera were tested with a panel of commercially available enzyme-linked immunosorbent assay kits based on recombinant forms of type VII collagen, and bullous pemphigoid antigens BP180 and BP230 (MBL, Nagoya, Japan). Sera that failed to react by enzyme-linked immunosorbent assay were analyzed by immunoblotting with dermal extract, as described elsewhere.[6],[11],[12]
Results
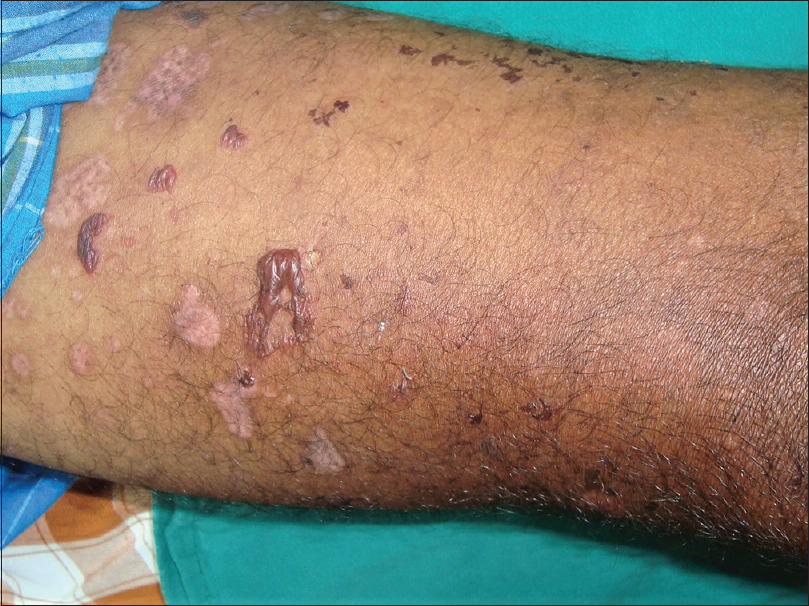
There were equal numbers of males and females in the study group and their mean age was 51.3 years (range, 11-77 years). All the patients showed linear deposition of immunoreactants (IgG and C3) along the basement membrane zone on direct immunofluorescence microscopy [Figure - 1]. The clinical profile of patients is summarised in [Table - 1]. Three patients (Nos. 2, 9, 10) presented with tense blisters on trauma-prone areas of the extremities and dorsa of hands and feet, which healed with scarring and milia formation [Figure 2a] and [Figure 2b]. This subgroup of patients was clinically diagnosed to have the classical mechanobullous phenotype of epidermolysis bullosa acquisita, compatible with a floor-binding indirect immunofluorescence pattern with salt-split skin. All other patients (Nos. 1, 3-8) presented with generalized tense blisters and erosions and were clinically diagnosed as having bullous pemphigoid initially [Figure - 3] and [Figure - 4]. However, since indirect immunofluorescence microscopy on salt-split skin showed an exclusive floor-binding pattern in all but one of them, a revised diagnosis of the “inflammatory” subtype of epidermolysis bullosa acquisita was made. One patient showed staining of both the epidermal and dermal sides of the split (floor > roof) on salt-split skin [Table - 2].
 |
| Figure 1: Anti-p200 pemphigoid showing linear deposits of immunoglobulin G at the basement membrane zone by direct immunofluorescence microscopy (fluorescein isothiocyanate, ×200) |
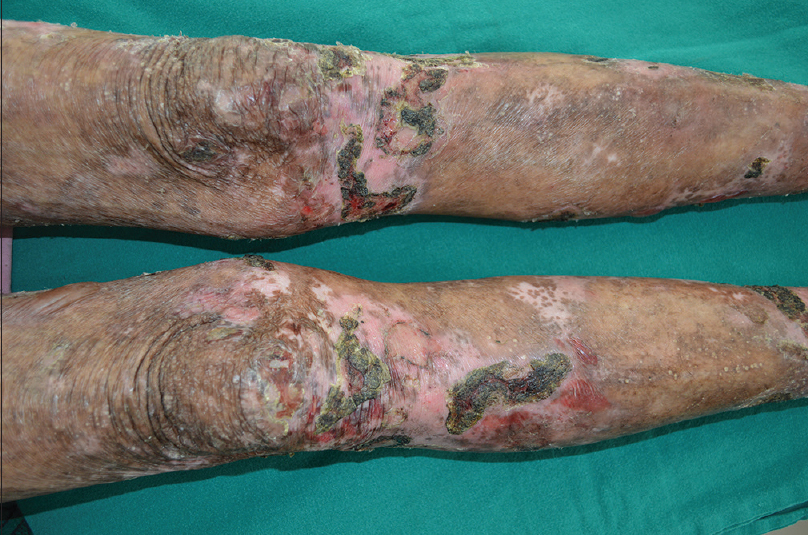 |
| Figure 2a: Classical mechanobullous epidermolysis bullosa acquisita: erosions, crusts, and milia on the knees and legs |
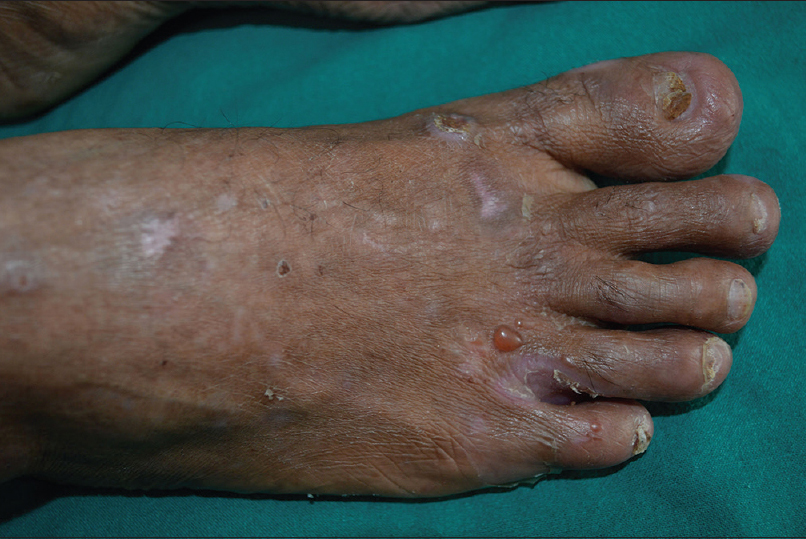 |
| Figure 2b: Epidermolysis bullosa acquisita showing tense blisters and nail dystrophy on the right foot |
 |
| Figure 3: Inflammatory epidermolysis bullosa acquisita: tense blisters on erythematous background on the left foot |
 |
| Figure 4: Anti-p200 pemphigoid: tense blisters, erosions, and crusts in the left cubital fossa |

Six patients (three each of the classical and inflammatory subtype) had absent basement membrane zone staining on recessive dystrophic epidermolysis bullosa skin [Figure - 5] but had circulating antibodies against type VII collagen by enzyme-linked immunosorbent assay, thus confirming the diagnosis of epidermolysis bullosa acquisita in them [Table - 2].
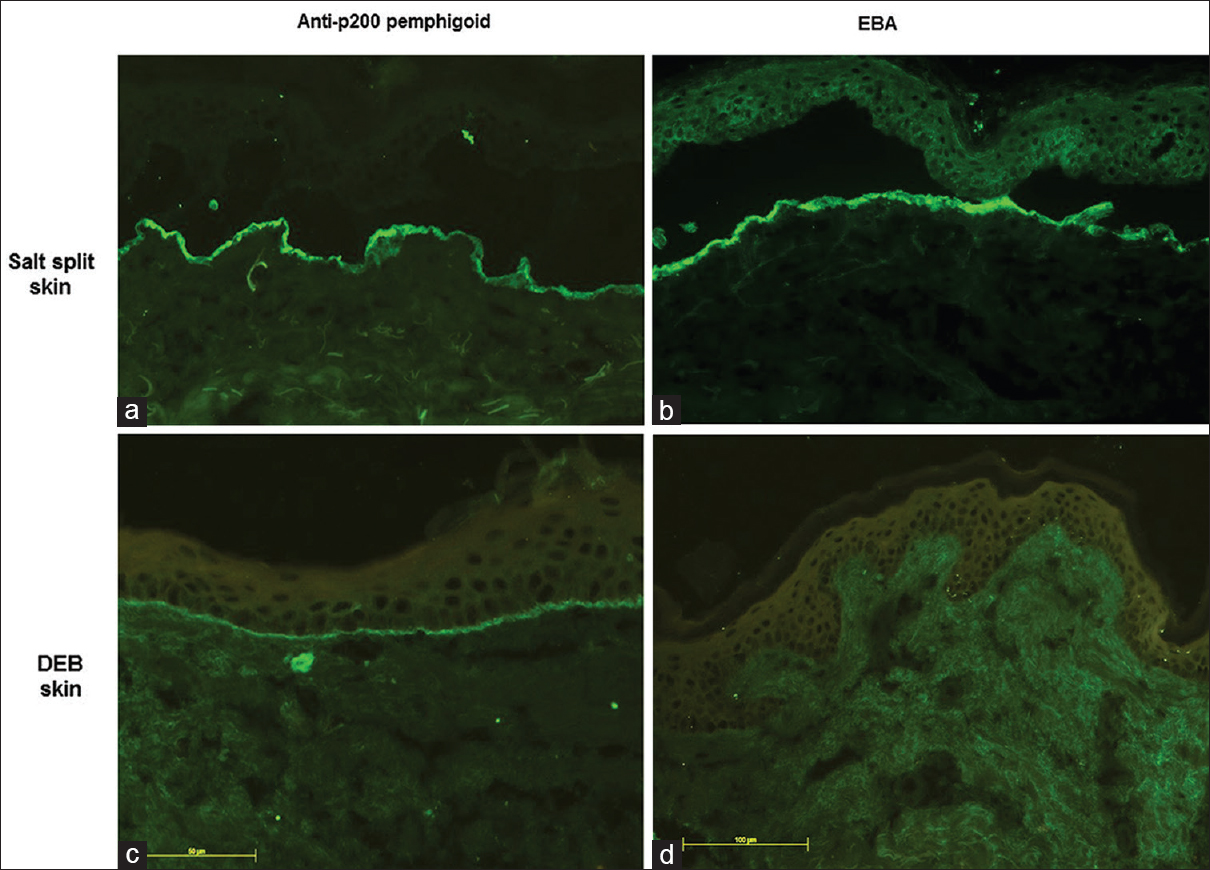 |
| Figure 5: Indirect immunofluorescence microscopy on salt-split skin showing floor binding pattern in anti-p200 pemphigoid (a) epidermolysis bullosa acquisita (b) Modified indirect immunofluorescence microscopy using patient's sera and recessive dystrophic epidermolysis bullosa skin as a substrate showing persistent basement membrane zone staining in anti-p200 pemphigoid (c) absence of basement membrane zone staining in epidermolysis bullosa acquisita (d) (fluorescein isothiocyanate, ×200) |
Four patients (Nos. 4, 5, 6, 7) showed basement membrane zone staining on recessive dystrophic epidermolysis bullosa skin [Figure - 5] and were negative on type VII collagen enzyme-linked immunosorbent assay, ruling out the diagnosis of epidermolysis bullosa acquisita. In three of these patients, immunoblotting with dermal extracts revealed immunoglobulin G4 reactivity against a 200 kDa protein [Figure - 6]. One of these four patients (No. 5) with basement membrane zone labeling on recessive dystrophic epidermolysis bullosa skin was found to have anti-bullous pemphigoid 180 antibodies by enzyme-linked immunosorbent assay.
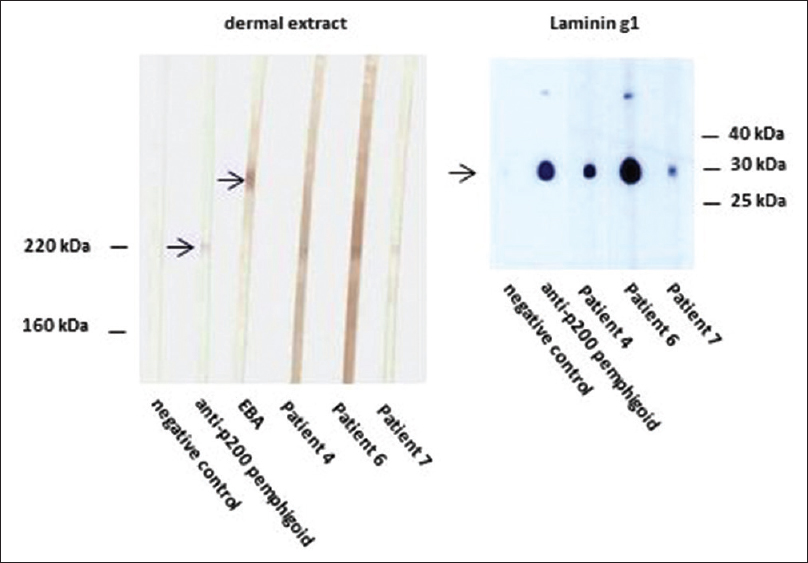 |
| Figure 6: Western blotting of sera with extract of human dermis (left panel). Like the serum of a control anti-p200 pemphigoid patient, sera from patients 4, 6, and 7 show reactivity with a 200 kDa molecule. Serum of a control epidermolysis bullosa acquisita (EBA) patient reacts with the 290 kDa type VII collagen. Western botting with recombinant laminin γ1 (right panel). Sera from patients 4, 6, and 7, like the control anti-p200 pemphigoid serum, react with this recombinant protein. Negative control sera show no reactivity in both panels |
Discussion
There is considerable overlap among various subepidermal autoimmune bullous diseases and their precise diagnosis can be a challenge. Most of these disorders present with tense blisters and erosions on the skin with or without mucous membrane involvement; direct immunofluorescence microscopy too may not reveal the diagnosis unequivocally.[13] Indirect immunofluorescence microscopy on salt-split skin is a simple and reliable tool which helps to sub classify subepidermal autoimmune bullous diseases into 'roof' and 'floor' binding conditions.[3]
Epidermolysis bullosa acquisita, a typical floor-binding subepidermal autoimmune bullous diseases, is characterized by the presence of immunoglobulin G autoantibodies directed against the noncollagenous amino-terminal (NC1) domain of type VII collagen, a major component of anchoring fibrils.[14] Two main presentations of epidermolysis bullosa acquisita are recognized; the “classic mechanobullous” and the “inflammatory vesiculobullous” types, the latter being twice as common as the former.[15] The mechanobullous phenotype is characterized by noninflammatory blisters on trauma-prone areas such as elbows, knees as well as the dorsa of hands and feet, which often heal with scarring and milia formation. The inflammatory form of epidermolysis bullosa acquisita manifests as widespread blisters with erythematous bases. Clinically, it may not be possible to distinguish the inflammatory form form of epidermolysis bullosa acquisita from other subepidermal autoimmune bullous diseases including bullous pemphigoid.[16] Since epidermolysis bullosa acquisita is more difficult to treat and has a poorer prognosis than other subepidermal autoimmune bullous diseases,[17] it is important to distinguish between these disorders so that early and appropriate treatment can be instituted.
Although indirect immunofluorescence microscopy on salt-split skin is a simple technique to diagnose subepidermal autoimmune bullous diseases, it cannot clearly differentiate between all of them. For example, floor binding of salt-split skin is not only found in epidermolysis bullosa acquisita but also in anti-laminin 332 mucous membrane pemphigoid and anti-p200 pemphigoid.[18] Subepidermal autoimmune bullous diseases with floor-binding autoantibodies against antigens that have not yet been identified have also been reported.[19],[20]
Serration pattern analysis of direct immunofluorescence microscopy may be highly valuable to distinguish epidermolysis bullosa acquisita ('u' serration pattern) from all other pemphigoid diseases ('n' serration pattern).[21],[22],[23] However, this technique is not yet widely available in the community and requires frozen sections of ideally 4-6 μm.
Immunomapping using a panel of skin deficient in certain basement membrane zone constituents is a fairly simple procedure that can be done in any laboratory with a facility for immunofluorescence microscopy. Compared to other techniques for the identification of the target antigen(s) such as immunoblotting and immunoprecipitation, immunomapping is less time-consuming and labor-intensive. Its main disadvantage however is the limited availability of suitable skin deficient in basement membrane zone components.[13]
Chen et al. first reported the utility of an enzyme-linked immunosorbent assay to detect type VII collagen autoantibodies in epidermolysis bullosa acquisita patients.[24] Subsequently, it has been shown that this test is useful not only for the diagnosis of epidermolysis bullosa acquisita but also in the evaluation of disease severity.[25],[26],[27] Several commercially available enzyme-linked immunosorbent assay systems based on the immunodominant NC1 domain of type VII collagen (alone or in combination with NC2) have been studied, with reported sensitivities of 66–100% and specificities of 98–100%.[26],[27] In our study, diagnosis of epidermolysis bullosa acquisita was made in six patients based on the results of both immunomapping using recessive dystrophic epidermolysis bullosa skin, and type VII collagen enzyme-linked immunosorbent assay.
Yet another tool, serration pattern analysis, appears to be a valuable and easily learnt method to differentiate autoimmunity against type VII collagen from other pemphigoid diseases by direct immunofluorescence microscopy.[21],[23] A u-serrated pattern (arches closed at the bottom) appears with epidermolysis bullosa acquisita and bullous systemic lupus erythematosus while an n-serration pattern (arches closed at the top) is seen in all other pemphigoid diseases. Pattern analysis of direct immunofluorescence microscopy samples could be included in the routine reporting of all subepidermal autoimmune bullous diseases. However this technique is not yet widely available and ideally requires frozen sections of 4–6 μm. One of our patients had mucocutaneous disease and circulating autoantibodies against the BP180 antigen. Serration pattern analysis by direct immunofluorescence microscopy and additional serological testing for the presence of anti-laminin 332 antibodies (to rule out mucous membrane pemphigoid) may have helped in confirming the diagnosis in this patient, but he was lost to follow-up and further testing could not be done.
Ours is the first report of cases with anti-p200 pemphigoid from India (n = 3). This condition was first described in 1996.[6],[28] Typically, it affects patients between 50 and 70 years of age, which is considerably less than the average age of patients with bullous pemphigoid.[9],[29] Clinically, anti-p200 pemphigoid is characterized by tense blisters on an urticarial background; hence, it can be easily mistaken for other subepidermal autoimmune bullous diseases, particularly bullous pemphigoid and the inflammatory variant of epidermolysis bullosa acquisita. Coexistence with psoriasis has been reported in 30% of cases, especially in Japanese patients.[28],[29] Histopathology of anti-p200 pemphigoid, with subepidermal blisters containing neutrophils and occasionally eosinophils, does not allow clear differentiation from other subepidermal autoimmune bullous diseases.[30] Direct immunofluorescence microscopy reveals a linear deposition of immunoglobulin G and C3 at the dermal-epidermal junction with an n-serrated pattern, similar to bullous pemphigoid. Indirect immunofluorescence microscopy on salt-split skin helps differentiate anti-p200 pemphigoid from bullous pemphigoid as anti-p200 pemphigoid stains the dermal side and bullous pemphigoid, the epidermal side of salt-split skin.[28] However, the above mentioned tests, except serration pattern analysis of direct immunofluorescence microscopy, are not useful to distinguish anti-p200 pemphigoid from epidermolysis bullosa acquisita. It is important to differentiate these two diseases as anti-p200 pemphigoid has a relatively benign course and can be treated with potent topical steroids, alone or in combination with oral dapsone.[7],[9]
Immunoblotting studies in anti-p200 pemphigoid using extracts of human dermis have revealed that the autoantibodies target a 200 kDa molecule.[6] Dainichi et al. showed that about 90% of patients' sera react with laminin γ1, a 200 kDa glycoprotein and they therefore proposed the term “anti-laminin γ1 pemphigoid” for this condition.[29] Laminin γ1 is present in the lower lamina lucida and contributes to dermal-epidermal adhesion outside hemidesmosomes. The C-terminus of laminin γ1 is recognized as an immunodominant region and autoantibodies may preferentially target this particular epitope.[12] Groth et al. confirmed these findings and established a novel enzyme-linked immunosorbent assay using a recombinant C-terminal fragment of laminin γ1 expressed in Escherichia coli with a sensitivity of 77% and a specificity of 99%.[11]
In contrast to some previous reports, the mean age of our patients with anti-p200 pemphigoid was 75 years and none of them had coexistent psoriasis. They all presented with tense blisters and erosions, predominantly over the extremities; one patient had predominant flexural involvement. Oral erosions were present in all three patients but did not dominate over skin lesions, which resolved without scarring or milia formation. Initially, in these three patients, a clinical diagnosis of bullous pemphigoid had been made which, in view of the floor-binding pattern on indirect immunofluorescence microscopy with salt-split skin, was subsequently revised to the inflammatory subtype of epidermolysis bullosa acquisita. However, persistent basement membrane zone labeling on recessive dystrophic epidermolysis bullosa skin and a negative type VII collagen enzyme-linked immunosorbent assay ruled out this diagnosis of epidermolysis bullosa acquisita as well. The final diagnosis of anti-p200 pemphigoid was established by immunoblotting with dermal extracts which showed reactivity with the p200 protein.
Conclusion
Indirect immunofluorescence microscopy on salt-split skin should be done as a first step in the differentiation of the subepidermal autoimmune bullous diseases. Immunomapping using a panel of epidermolysis bullosa skin samples or, if available, immunoblotting and a specialized enzyme-linked immunosorbent assay can be used to identify target antigens. The diagnosis of anti-p200 pemphigoid may be suspected in patients with floor binding on indirect immunofluorescence microscopy with salt-split skin, basement membrane zone staining of type VII collagen-deficient skin, and negative type VII collagen enzyme-linked immunosorbent assay reactivity. Diagnosis of anti-p200 pemphigoid is confirmed by the detection of serum autoantibodies against the p200 protein by immunoblotting with dermal extracts and/or recombinant laminin γ1.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Schmidt E, Zillikens D. Pemphigoid diseases. Lancet 2013;381:320-32.
[Google Scholar]
|
| 2. |
Schmidt E, Zillikens D. The diagnosis and treatment of autoimmune blistering skin diseases. Dtsch Arztebl Int 2011;108:399-405.
[Google Scholar]
|
| 3. |
Lazarova Z, Yancey KB. Reactivity of autoantibodies from patients with defined subepidermal bullous diseases against 1 mol/L salt-split skin. Specificity, sensitivity, and practical considerations. J Am Acad Dermatol 1996;35(3 Pt 1):398-403.
[Google Scholar]
|
| 4. |
Domloge-Hultsch N, Anhalt GJ, Gammon WR, Lazarova Z, Briggaman R, Welch M, et al. Antiepiligrin cicatricial pemphigoid. A subepithelial bullous disorder. Arch Dermatol 1994;130:1521-9.
[Google Scholar]
|
| 5. |
Hashimoto T, Murakami H, Senboshi Y, Kanzaki H, Arata J, Yancey KB, et al. Antiepiligrin cicatricial pemphigoid: The first case report from Japan. J Am Acad Dermatol 1996;34(5 Pt 2):940-2.
[Google Scholar]
|
| 6. |
Zillikens D, Kawahara Y, Ishiko A, Shimizu H, Mayer J, Rank CV, et al. Anovel subepidermal blistering disease with autoantibodies to a 200-kDa antigen of the basement membrane zone. J Invest Dermatol 1996;106:1333-8.
[Google Scholar]
|
| 7. |
Dilling A, Rose C, Hashimoto T, Zillikens D, Shimanovich I. Anti-p200 pemphigoid: A novel autoimmune subepidermal blistering disease. J Dermatol 2007;34:1-8.
[Google Scholar]
|
| 8. |
Dainichi T, Kurono S, Ohyama B, Ishii N, Sanzen N, Hayashi M, et al. Anti-laminin gamma-1 pemphigoid. Proc Natl Acad Sci U S A 2009;106:2800-5.
[Google Scholar]
|
| 9. |
Goletz S, Hashimoto T, Zillikens D, Schmidt E. Anti-p200 pemphigoid. J Am Acad Dermatol 2014;71:185-91.
[Google Scholar]
|
| 10. |
Vodegel RM, Kiss M, Cjm De Jong M, Pas HH, Altmayer A, Molnar K, et al. The use of skin substrates deficient in basement membrane molecules for the diagnosis of subepidermal autoimmune bullous disease. Eur J Dermatol 1998;8:83-5.
[Google Scholar]
|
| 11. |
Groth S, Recke A, Vafia K, Ludwig RJ, Hashimoto T, Zillikens D, et al. Development of a simple enzyme-linked immunosorbent assay for the detection of autoantibodies in anti-p200 pemphigoid. Br J Dermatol 2011;164:76-82.
[Google Scholar]
|
| 12. |
Vafia K, Groth S, Beckmann T, Hirose M, Dworschak J, Recke A, et al. Pathogenicity of autoantibodies in anti-p200 pemphigoid. PLoS One 2012;7:e41769.
[Google Scholar]
|
| 13. |
Rao R, Bhogal B, Groves R. Antigen identification using skin deficient in basement-membrane protein: A novel tool for the diagnosis of subepidermal immunobullous diseases. Clin Exp Dermatol 2013;38:289-94.
[Google Scholar]
|
| 14. |
Lapiere JC, Woodley DT, Parente MG, Iwasaki T, Wynn KC, Christiano AM, et al. Epitope mapping of type VII collagen. Identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J Clin Invest 1993;92:1831-9.
[Google Scholar]
|
| 15. |
Buijsrogge JJ, Diercks GF, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol 2011;165:92-8.
[Google Scholar]
|
| 16. |
Gupta R, Woodley DT, Chen M. Epidermolysis bullosa acquisita. Clin Dermatol 2012;30:60-9.
[Google Scholar]
|
| 17. |
Calabresi V, Sinistro A, Cozzani E, Cerasaro C, Lolicato F, Muscianese M, et al. Sensitivity of different assays for the serological diagnosis of epidermolysis bullosa acquisita: Analysis of a cohort of 24 Italian patients. J Eur Acad Dermatol Venereol 2014;28:483-90.
[Google Scholar]
|
| 18. |
Meijer JM, Diercks GF, Schmidt E, Pas HH, Jonkman MF. Laboratory diagnosis and clinical profile of Anti-p200 pemphigoid. JAMA Dermatol 2016;152:897-904.
[Google Scholar]
|
| 19. |
Chan LS, Fine JD, Briggaman RA, Woodley DT, Hammerberg C, Drugge RJ, et al. Identification and partial characterization of a novel 105-kDalton lower lamina lucida autoantigen associated with a novel immune-mediated subepidermal blistering disease. J Invest Dermatol 1993;101:262-7.
[Google Scholar]
|
| 20. |
Allen J, Wojnarowska F. Linear IgA disease: The IgA and IgG response to dermal antigens demonstrates a chiefly IgA response to LAD285 and a dermal 180-kDa protein. Br J Dermatol 2003;149:1055-8.
[Google Scholar]
|
| 21. |
Vodegel RM, Jonkman MF, Pas HH, de Jong MC. U-serrated immunodeposition pattern differentiates type VII collagen targeting bullous diseases from other subepidermal bullous autoimmune diseases. Br J Dermatol 2004;151:112-8.
[Google Scholar]
|
| 22. |
Terra JB, Jonkman MF, Diercks GF, Pas HH. Low sensitivity of type VII collagen enzyme-linked immunosorbent assay in epidermolysis bullosa acquisita: Serration pattern analysis on skin biopsy is required for diagnosis. Br J Dermatol 2013;169:164-7.
[Google Scholar]
|
| 23. |
Terra JB, Meijer JM, Jonkman MF, Diercks GF. The n- vs. u-serration is a learnable criterion to differentiate pemphigoid from epidermolysis bullosa acquisita in direct immunofluorescence serration pattern analysis. Br J Dermatol 2013;169:100-5.
[Google Scholar]
|
| 24. |
Chen M, Chan LS, Cai X, O'Toole EA, Sample JC, Woodley DT. Development of an ELISA for rapid detection of anti-type VII collagen autoantibodies in epidermolysis bullosa acquisita. J Invest Dermatol 1997;108:68-72.
[Google Scholar]
|
| 25. |
Saleh MA, Ishii K, Kim YJ, Murakami A, Ishii N, Hashimoto T, et al. Development of NC1 and NC2 domains of type VII collagen ELISA for the diagnosis and analysis of the time course of epidermolysis bullosa acquisita patients. J Dermatol Sci 2011;62:169-75.
[Google Scholar]
|
| 26. |
Komorowski L, Müller R, Vorobyev A, Probst C, Recke A, Jonkman MF, et al. Sensitive and specific assays for routine serological diagnosis of epidermolysis bullosa acquisita. J Am Acad Dermatol 2013;68:e89-95.
[Google Scholar]
|
| 27. |
Kim JH, Kim YH, Kim S, Noh EB, Kim SE, Vorobyev A, et al. Serum levels of anti-Col VII antibodies detected by enzyme-linked immunosorbent assay in patients with epidermolysis bullosa acquisita are correlated with the severity of skin lesions. J Eur Acad Dermatol Venereol 2013;27:224-30.
[Google Scholar]
|
| 28. |
Chen KR, Shimizu S, Miyakawa S, Ishiko A, Shimizu H, Hashimoto T. Coexistence of psoriasis and an unusual IgG-mediated subepidermal bullous dermatosis: Identification of a novel 200-kDa lower lamina lucida target antigen. Br J Dermatol 1996;134:340-6.
[Google Scholar]
|
| 29. |
Dainichi T, Koga H, Tsuji T, Ishii N, Ohyama B, Ueda A, et al. From anti-p200 pemphigoid to anti-laminin gamma1 pemphigoid. J Dermatol 2010;37:231-8.
[Google Scholar]
|
| 30. |
Rose C, Weyers W, Denisjuk N, Hillen U, Zillikens D, Shimanovich I. Histopathology of anti-p200 pemphigoid. Am J Dermatopathol 2007;29:119-24.
[Google Scholar]
|
Fulltext Views
4,839
PDF downloads
2,430





![[Figure - 1]](#fig_ijdvl_2017_83_5_550_211723_f1.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2017_83_5_550_211723_t7.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2017_83_5_550_211723_f5.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2017_83_5_550_211723_f6.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2017_83_5_550_211723_t8.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2017_83_5_550_211723_f7.jpg){kind=link}
![[Figure - 6]](#fig_ijdvl_2017_83_5_550_211723_f8.jpg){kind=link}