Translate this page into:
Familial angiokeratoma corporis diffusum without identified enzyme defect
2 Faculty of Medicine, School of Medicine, National Yang-Ming University, Taipei, Taiwan
3 Division of Neurosurgery, Department of Surgery, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung; Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
Correspondence Address:
Chieh-Hsin Wu
No. 100, Tzyou 1st Road, Kaohsiung City 807
Taiwan
| How to cite this article: Lu YY, Lu CC, Wu CS, Wu CH. Familial angiokeratoma corporis diffusum without identified enzyme defect. Indian J Dermatol Venereol Leprol 2015;81:46-49 |
Abstract
Angiokeratoma corporis diffusum is the cutaneous hallmark of several rare inherited lysosomal diseases associated with specific enzyme deficiencies in the metabolism of glycoproteins, most notably Fabry disease. These defects result in many systemic manifestations. Here, we report a rare familial case of angiokeratoma corporis diffusum that developed at puberty with no major systemic manifestations and no underlying enzyme defect or gene mutation. Familial angiokeratoma corporis diffusum without identified enzyme defect appears to be a distinct clinical entity with a benign course.INTRODUCTION
Angiokeratomas are classified into either localized or diffuse forms. The localized form includes solitary papular angiokeratoma, angiokeratoma of Fordyce, angiokeratoma of Mibelli, and angiokeratoma circumscriptum neviforme. The diffuse form is angiokeratoma corporis diffusum which is systemic. In contrast with solitary angiokeratoma, diffuse angiokeratomas should alert the physician to determine whether the patient has an underlying lysosomal storage disease or other systemic manifestations. Vary rarely, angiokeratoma corporis diffusum have been reported in patients with normal enzyme activity. In the familial case reported here, the patient not only had normal enzyme activity but also showed no evidence of underlying systemic abnormalities. Other reports of angiokeratoma corporis diffusum with normal enzyme function are also reviewed. We hypothesize that angiokeratoma corporis diffusum is always related to a known or unknown enzymatic defect such as an enzymatic polymorphism though the enzyme activity may be in the normal range. [1]
Case Report
A 26-year-old Taiwanese woman complained of multiple discrete erythematous to dark-red non-blanchable papules that began developing over her left buttock as a teenager and then progressed to the right buttock [Figure - 1]. These eruptions coalesced and became increasingly numerous and hyperkeratotic with age. Her 24-year-old sister also had similar skin lesions with multiple discrete dark-red papules on the right buttock with progression to the right thigh [Figure - 2]. In both patients, the face was unaffected, and intelligence was normal. Neither complained of systemic symptoms such as pain in the extremities, nausea, vomiting, hypohidrosis, frothy urine, hearing disturbance, fever, headache, chest pain, or dyspnea. In addition, neither patient had a history of metabolic disorders; renal, cardiac, or neurological disease; or delayed puberty. Other than the skin eruptions in the bathing trunk area, physical examination was normal. There was no history of trauma. Dermoscopy revealed sharply-demarcated groups of red lacunae. Skin biopsy of the lesions showed overlying compact orthrokeratotic, acanthotic epidermis encircling superficial ectatic capillaries lined with normal-appearing endothelial cells [Figure - 3], which was consistent with angiokeratoma. Electron microscopy showed normal endothelial cells without lamellated intracytoplasmic vacuolar inclusions or zebra bodies. The patients were diagnosed with angiokeratoma corporis diffusum. No other family member had the disease or a relevant clinical history [Figure - 4]. Enzyme activity, including α-galactosidase A (Fabry disease), α-N-acetylgalactosaminidase (Kanzaki disease), β-mannosidase (β-mannosidosis), β-galactosidase (GM1 gangliosidosis), and α-fucosidase (fucosidosis), aspartylglucosaminidase (aspartylglucosaminuria), neuraminidase (sialidosis) were within normal limits in addition to gene mutation analysis of Fabry disease. Hemogram, blood biochemistry panel (including serum urea nitrogen and creatinine level), and liver function were all normal. Urinalysis, ophthalmologic slit-lamp examination, audiometry, chest X-ray, spirometry, and renal and cardiac ultrasonography were unremarkable. A Doppler echocardiogram of soft tissues showed no underlying arterial or venous malformation over the subcutaneous region of the bilateral buttocks and no abnormalities in the bilateral femoral or popliteal arteries or veins. Based on these results, they were diagnosed as familial angiokeratoma corporis diffusum without associated metabolic disease.
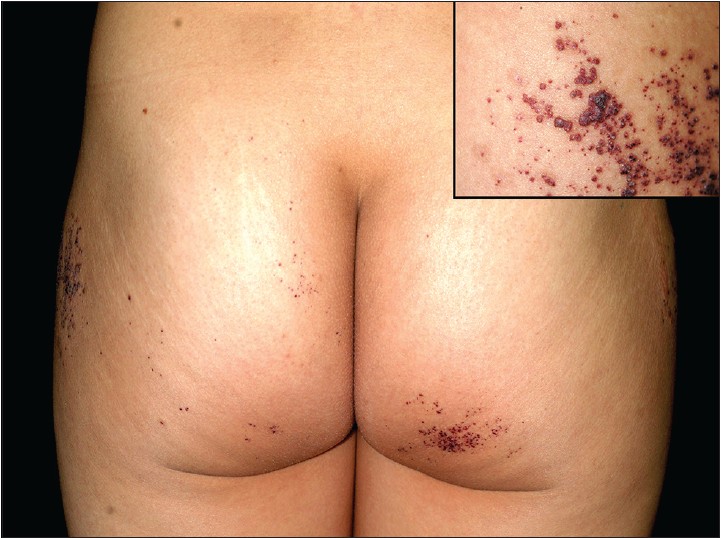 |
| Figure 1: Multiple discrete erythematous to dark-red non-blanchable papules in classic "bathing trunk" distribution. The inset shows coalescent hyperkeratotic plaques |
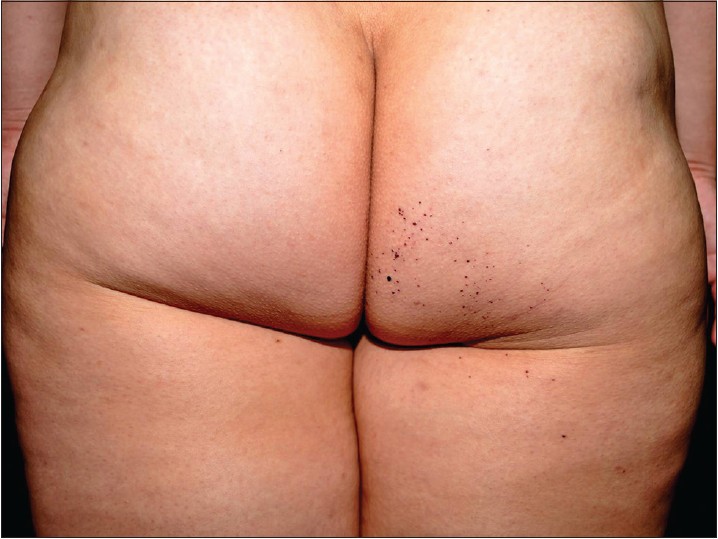 |
| Figure 2: Multiple discrete dark-red papules on the right buttock and thigh of the younger sister |
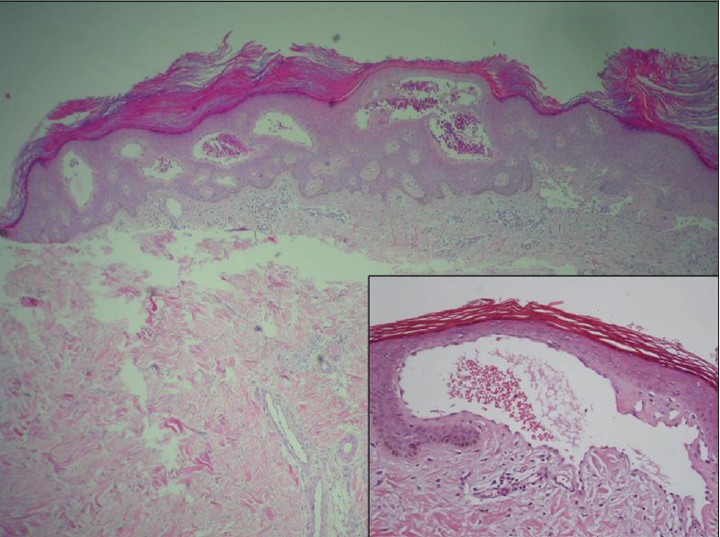 |
| Figure 3: Histopathological examination showing overlying compact orthrokeratosis, acanthosis and superficial ectatic capillaries lined with normal-appearing endothelial cells (H and E, ×40 and ×100). The inset shows dilated blood vessels with congested thin walls |
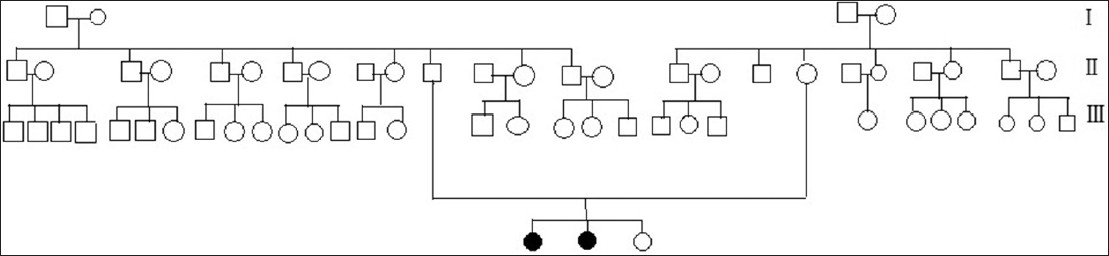 |
| Figure 4: Three-generation family pedigree. The black symbol indicates that the patient was affected by angiokeratoma corporis diffusum |
DISCUSSION
The term "angiokeratoma" is derived from the Greek words for vessel, horn, and tumor. [2] This benign capillary vascular malformation is quite common and is characterized by red papules which are initially soft and later become warty and keratotic. Angiokeratomas can be classified as localized or systemic. Solitary or localized forms are acquired, non-genetic disorders. Unlike the more common systemic forms, solitary forms are unassociated with underlying systemic disease. [3],[4]
Angiokeratomas are classified into five types according to their clinical characteristics. [2] The localized form includes: (1) The solitary papular type, which can manifest on any body part as a single lesion; (2) the Fordyce type, which occurs on the scrotum or the vulva; (3) the Mibelli type, which presents as hyperkeratotic vascular lesions over the bony prominences of the hands and feet; and (4) angiokeratoma circumscriptum neviforme, which is a congenital unilateral zosteriform verrucous lesion of the lower extremities. The systemic form, angiokeratoma corporis diffusum, can involve the entire body and is associated with lysosomal storage disease. [3]
Widespread angiokeratomas are diffuse and manifest as clustered symmetrical papules on the "bathing trunk" area of the body. Angiokeratoma corporis diffusum is a prominent cutaneous feature of lysosomal storage disease, including Fabry disease, GM1 gangliosidosis, aspartylglucosaminuria, fucosidosis, β-mannosidosis, sialidosis, galactosialidosis, and Kanzaki disease. Idiopathic cases have also been reported. [3],[5] Each of these storage diseases involves a distinctive enzyme deficiency. Apart from X-linked Fabry disease, all of these lysosomal enzyme defects are inherited in an autosomal recessive fashion, and all are characterized by some degree of central nervous system dysfunction, a characteristic facies, and often mental retardation and organomegaly. Electron microscope studies show that, unlike the electron-dense, lamellar (zebra-like) inclusions in endothelial and other cell types characteristic of Fabry disease, electron-lucent lysosomal dilation occurs in other lysosomal storage diseases. [1],[6]
A literature review showed 20 case reports of angiokeratoma corporis diffusum in the absence of metabolic disorder, and only one other familial case has been reported. [7] Reports are almost evenly split between male and female patients, and most patients are younger than 40 years. Familial cases tend to involve younger females. [8] No cases have revealed impaired intelligence or systemic vascular events, and the first appearance of the lesions is always during puberty. However, the first hospital visits are made an average of 10.8 years after onset of the eruption, which implies that the severity of disease gradually increases over time. The lower body is the most commonly affected area in the reported familial cases as was seen in our cases. None of the reported cases revealed lysosomal inclusions on electron microscopy of biopsy specimens. [1],[4],[5],[7],[8],[9],[10] Not all cases underwent a complete survey of enzyme activity for related lysosomal storage disease. Gene mutational analysis was only performed in two reported cases. [1],[7] Our case received complete workup for all related lysosomal storage disease and gene mutational analysis.
Familial occurrence strongly suggests a hereditary disease. Further studies may reveal other enzyme abnormalities associated with angiokeratoma corporis diffusum. Molecular biology studies of otherwise healthy patients with angiokeratoma corporis diffusum are needed to investigate whether the genetic profile or enzyme alterations affect the development of angiokeratomas. Since enzyme therapies have been developed for treating Fabry disease, it is important to be aware that other diseases can cause angiokeratoma corporis diffusum. [10] For example, angiokeratoma corporis diffusum in the absence of metabolic disease may be related to an unknown enzymatic or isoenzymatic defect. In addition, the possibility that angiokeratoma corporis diffusum is linked to a genetic and phenotypic polymorphism of a lysosomal storage disease such as Fabry disease has not been excluded. [5] Therefore careful observation of the clinical course and continuous accumulation of information on this enzymatic disorder is required. [7]
| 1. |
Kelly B, Kelly E. Angiokeratoma corporis diffusum in a patient with no recognizable enzyme abnormalities. Arch Dermatol 2006;142:615-8.
[Google Scholar]
|
| 2. |
Mittal R, Aggarwal A, Srivastava G. Angiokeratoma circumscriptum: A case report and review of the literature. Int J Dermatol 2005;44:1031-4.
[Google Scholar]
|
| 3. |
Mehta AB, Orteu CH. Fabry disease. In: Goldsmith LA, Fitzpatrick TB, editors. Fitzpatrick's Dermatology in General Medicine. 8 th ed. New York: McGraw-Hill; 2012. p. 1613 - 24.
th ed. New York: McGraw-Hill; 2012. p. 1613 - 24.'>[Google Scholar]
|
| 4. |
Calzavara-Pinton PG, Colombi M, Carlino A, Zane C, Gardella R, Clemente M, et al. Angiokeratoma corporis diffusum and arteriovenous fistulas with dominant transmission in the absence of metabolic disorders. Arch Dermatol 1995;131:57-62.
[Google Scholar]
|
| 5. |
Fimiani M, Mazzatenta C, Rubegni P, Andreassi L. Idiopathic angiokeratoma corporis diffusum. Clin Exp Dermatol 1997;22:205-6.
[Google Scholar]
|
| 6. |
Kanzaki T, Yokota M, Mizuno N, Matsumoto Y, Hirabayashi Y. Novel lysosomal glycoaminoacid storage disease with angiokeratoma corporis diffusum. Lancet 1989;1:875-7.
[Google Scholar]
|
| 7. |
Yasuike R, Nakai N, Komori S, Katoh N. Angiokeratoma corporis diffusum in the absence of metabolic disorders: A case report and mini-review of the published work. J Dermatol 2013;40:668-9.
[Google Scholar]
|
| 8. |
Lipsker D, Kieffer C, Nojavan H, Doray B, Cribier B. Familial ACD with no recognizable enzyme abnormalities. Arch Dermatol 2006;142:1509.
[Google Scholar]
|
| 9. |
Holmes RC, Fensom AH, McKee P, Cairns RJ, Black MM. Angiokeratoma corporis diffusum in a patient with normal enzyme activities. J Am Acad Dermatol 1984;10:384-7.
[Google Scholar]
|
| 10. |
Crovato F, Rebora A. Angiokeratoma corporis diffusum and normal enzyme activities. J Am Acad Dermatol 1985;12:885-6.
[Google Scholar]
|
Fulltext Views
5,235
PDF downloads
3,354





![[Figure - 1]](#fig_ijdvl_2015_81_1_46_148568_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_1_46_148568_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_1_46_148568_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2015_81_1_46_148568_f4.jpg){kind=link}