Translate this page into:
Fanconi anaemia
Correspondence Address:
D Masthan Saheb
Department of Dermatology, Govt. General Hospital, Kurnool - 518 002, Andhra Pradesh
India
| How to cite this article: Masthan Saheb D. Fanconi anaemia. Indian J Dermatol Venereol Leprol 2002;68:46-47 |
Abstract
A case of Fanconi anaemia in a 7 - year-old male child is reported. He was grossly anaemic with typical cutaneous pigmentary changes. Peripheral smear revealed normocytic hypochromic anaemia, leukopenia and thrombocytopenia. Abdominal ultrasonography revealed absence of right kidney.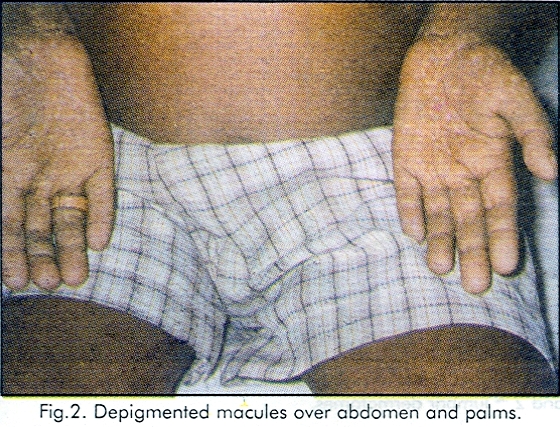 |
|
 |
|
Introduction
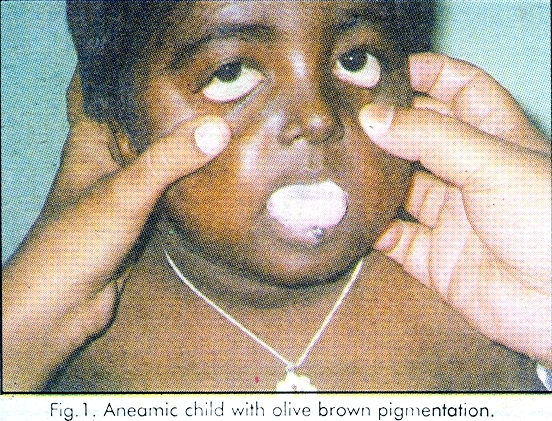
Fanconi anaemia (FA) is an autosomal recessive disease with progressive loss of all the formed elements of blood (pancytopenia) with generalised dusky or olive brown pigmentation particularly of the lower trunk, flexures and neck, superimposed with scattered rain drop - like depigmented and hyperpigmented macules. Occasionally cafe- au-lait spots are also present. These patients will also have skeletal malformations in the form of aplasia or hypoplasia of the thumbs, metacarpals or radius, hip dislocations or scoliosis,[1] Malformations of kidneys in the form of renal aplasia or horse shoe kidney, ocular abnormalities in the form of strabismus, microphthalmia and hypogonadism may be present. Some of these patients can also have hyperreflexia and mental retardation. These patients will have high incidence of neoplasias particularly nonlymphatic leukemia and hepatomas.[1] The course is often progressively downhill with death from infections, hemorrhages or neoplasia. Initially bone marrow function can be stimulated with corticosteroids and with androgenic steroid - oxymethalone.[2] Histocompatible bone marrow transplantation considerably improved survival period. The condition was first described by swiss paediatrician Guido Fanconi in 1927.[3]
Case Report
A 7 - year -old male child was brought to the department of Dermatology for asymptomatic hypopigmented lesions over the body since 5 - years. Physical examination revealed that he was grossly anaemic, and the skin was olive brown with symmetrical multiple discrete hypopigmented macules scattered all over the body especially on the neck, lower trunk, and on both palms. There were no cafe-au-lait spots. All the fingers including thumb were intact. Both the testes were present and were of normal size. There was no squint. His intelligence was normal. There was no palmoplantar kerotoderma. The nails were normal. All the superficial and deep reflexes were normal. There were no urinary abnormalities. Fundoscopy was normal. Peripheral smear revealed normocytic hypochromic anaemia, leukopenia and thrombocytopaenia. X-ray of hands was normal. Ultrasound examination of the abdomen revealed absence of kidney on right side. Bone marrow study and liver biopy could not be done because of thrombocytopaenia. There was no family history of similar, dermatological disorder. [Figure - 1], [Figure - 2]
Discussion
Fanconi anaemia is a rare disorder with incidence of 1 per 3,60,000 live births.[3] The diagnosis of FA is made by typical cutaneous and haematological findings. Cutaneous manifestations include generalised olive-brown pigmentation particularly on the neck, trunk and flexures superimposed with rain drop-like depigmented macules. The present case was having these macules even on both the palms though it was not described previously. Following repeated blood transfusions hyperpigmentation due to iron overload may be present.[1] Haematological manifestations include pancytopenia. Though cafe-au-lait spots were also described, no such spots were seen in the present case. Malformation of the kidneys in the form of aplasia or horse shoe kidney were described. Right kidney was absent in the present case. Though skeletal and opthalmological abnormalities, hypogondism, hyper reflexia, mental retardation, nonlymphatic leukemia, and hepatomas were described, the present case was not having any of these abnormalities. The only condition which will have these cutaneous changes with pancytopenia is dyskeratosis congenita. But early age of onset of hyperpigmentation, absence of palmoplantar hyperkeratosis, and renal involvement in the present case favour the diagnosis Fanconi anaemia.
| 1. |
Kenneth H. Kraemer. Heritable diseases with increased sensitivity to cellular injury, In: Fitzspatrick's Dermatology in General Medicine. Edited by Irwin M. Freedberg, Arthur Z. Eisen, 5th edn, New York. Mc Grow-Hill Health Profession division. 1999; 1857.
[Google Scholar]
|
| 2. |
Atherton DJ. The neonate, in: Textbook of Dermatology. Edited by Champion RH, Burton JL, Burns DA, 6th edn, Oxford. Blackwell Scientific Publications, 1998: 505 -506.
[Google Scholar]
|
| 3. |
Andrew Deiss. Non neoplastic diseases, chemical agents and haemolytic disorders that may precede haematology neoplasms. In: Wintrobes Clinical Hermatology. Edited by Richard Leeg, Thomas C. Bithell, 9th edn, London. Lea and Febiger. 1993; 1947.
[Google Scholar]
|





![[Figure - 1]](#fig_ijdvl_2002_68_1_46_12869_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2002_68_1_46_12869_2.jpg){kind=link}