
Translate this page into:
Gaucher disease with severe congenital ichthyosiform erythroderma in two siblings
Corresponding author: Dr. Aanchal Bansal, Department of Dermatology, Venereology and Leprosy, Lady Hardinge Medical College and Hospital, New Delhi, India. aanchal.bansal.ab125@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Mendiratta V, Bansal A, Jain A. Gaucher disease with severe congenital ichthyosiform erythroderma in two siblings. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_1176_2024
Dear Editor,
Gaucher disease (GD) is a rare, autosomal recessive, lysosomal disorder caused by the accumulation of glucosylceramides, resulting in diverse clinical presentations. We report two young Indian siblings with severe congenital ichthyosis and systemic complications related to type 2 GD.
A three-year-old child (patient 1) presented with peeling skin for 1-month of age, along with a history of seizures, fever, loose stools, decreased feeding and activity, frequent hospitalisations due to recurrent infections, multiple blood transfusions and failure to thrive. There was no history of collodion membrane at birth and consanguinity in the parents. The developmental milestones were delayed.
On general examination, the child had pallor, coarse facies, microcephaly, stunting and severe malnutrition. Cutaneous examination revealed ichthyosiform scaling, generalised desquamation and fissuring of skin over the whole body. Nails were thickened and dystrophic. Oral and genital mucosae were normal. Clinical examination suggested congenital ichthyosiform erythroderma (CIE) [Figures 1a and 1b].
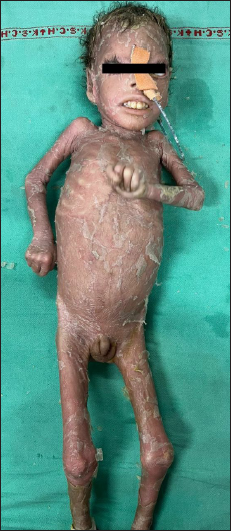
- Patient 1, Generalised ichthyosis with desquamation and fissuring and arthrogryposis.
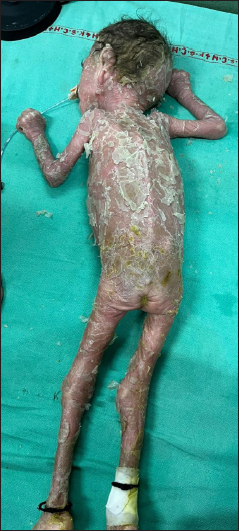
- Ichthyosiform, yellow-brown coarse scales with desquamation of skin over the back.
Ophthalmological examination revealed ectropion and bilateral corneal opacities [Figure 1c]. Musculoskeletal examination revealed arthrogryposis, hypotonia and spasticity. The remaining systemic examination was normal. Hepatosplenomegaly was absent. The patient had reduced haemoglobin (4.8 g/dl) with normal liver/kidney function tests. Magnetic resonance imaging (MRI) of the brain showed subacute haemorrhaging in bilateral ventricles along with pontocerebellar hypoplasia. Whole abdomen ultrasonography was unremarkable. Histopathological examination of the skin was consistent with CIE [Figure 2a, 2b]. Whole genomic sequencing of the patient’s blood sample depicted heterozygous missense variation (c.1448T>C) in exon 10 of the glucocerebrosidase (GBA) gene that led to substitution from leucine to proline at codon 483 (p.Leu483Pro), likely pathogenic variant suggestive of GD.
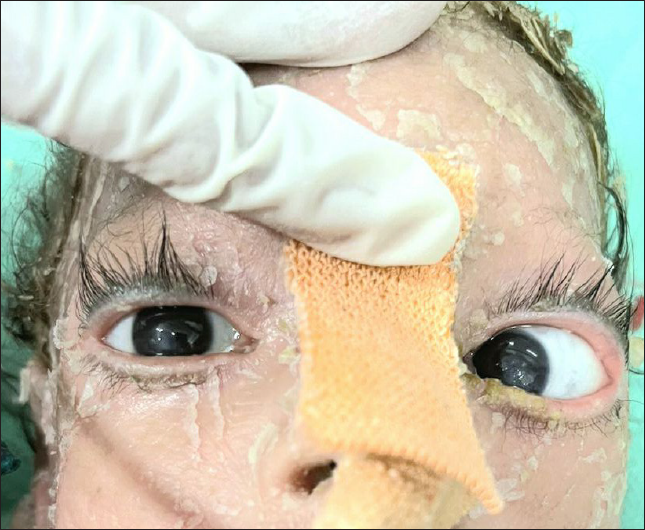
- Ectropion and visible corneal opacities in bilateral eyes.
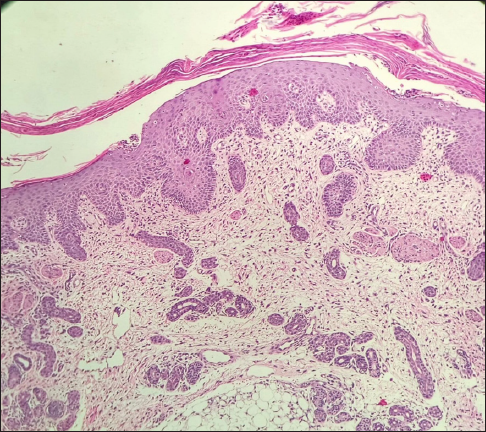
- Histopathological examination (Haematoxylin & eosin, 100×) shows hyperkeratosis and irregular acanthosis of the epidermis. Dermis shows mild perivascular lymphocytic infiltrate with melanin incontinence.
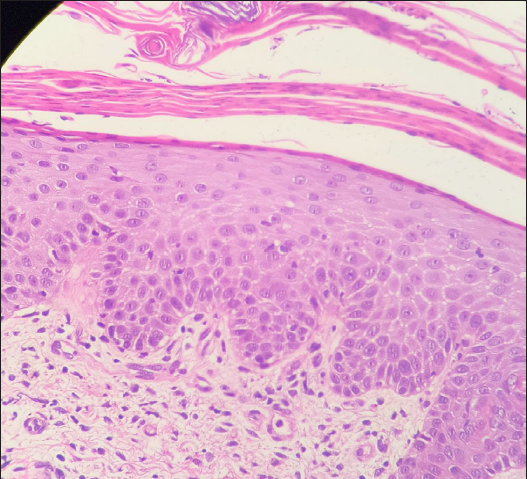
- Histopathological examination (Haematoxylin & eosin, 400×) shows lamellar hyperkeratosis with parakeratosis and exocytosis of neutrophils and lymphocytes.
The younger sibling of the above patient (patient 2), a three-month-old female, presented with similar cutaneous and systemic complaints since 1.5 months of age along with fever, cough, feeding difficulties and delayed developmental milestones. On general examination, pallor, microcephaly and severe stunting were evident. Cutaneous findings were similar to the elder child [Figures 3a and 3b]. Abdominal palpation revealed hepatomegaly 3 cm below and splenomegaly 2 cm below the respective costal margins. Musculoskeletal examination revealed reduced power (MRC grading 3/5) in bilateral upper and lower limbs with hypotonia. The rest of the systemic examination was normal. Investigations revealed reduced haemoglobin (6.8 g/dl), raised total leucocyte count (26,000/mm3) and hypoproteinaemia. Other investigations were normal. The skin biopsy findings were consistent with CIE. The younger child expired after a few days due to aspiration pneumonia and the older sibling was started on treatment with systemic antibiotics, topical emollients and antibiotic ointments. Parents were advised genetic counselling for the disease.
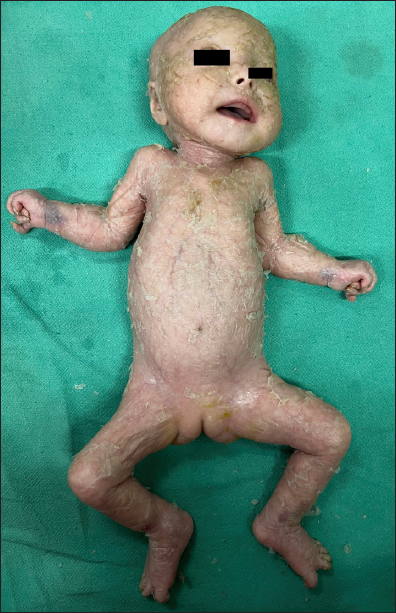
- Patient 2 – Generalised lamellar ichthyosis with fissuring of skin in folds, flexion contractures of upper and lower limbs, arthrogryposis and palmoplantar keratoderma.
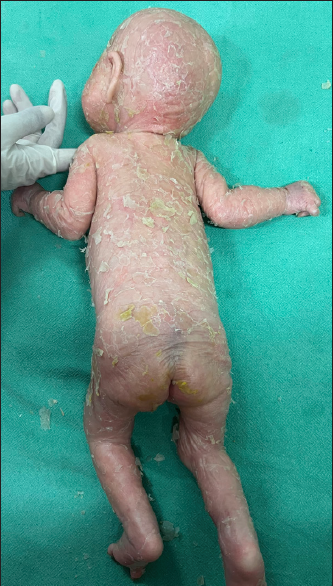
- Patient 2 – ichthyotic scaling, desquamation of skin over lower limbs.
GD is the most common lysosomal storage disorder resulting from deficiency of the GBA enzyme due to mutation of the GBA1 gene, located on chromosome 1 (1q21).1 There are three subsets of the disease. Type 1 GD is the non-neuronopathic form, while type 2 GD and type 3 GD are the neuronopathic forms.2 The phenotypes of GD are summarised in Table 1. Type 2 GD (acute, infantile form) is a rare sub-type of GD. It usually appears within two years of age, progresses rapidly, and death may occur by 3–4 years of age.3 The perinatal lethal (PNL) form of type 2 GD Table 1 is associated with onset in-utero, extending to 3 months of age and may lead to death within 3 months. Our 2nd patient presented with similar findings. The non-perinatal lethal form of type 2 GD presents relatively late, till the age of two years with visceral and/or neurologic symptoms and microscopic skin changes (immature lamellar membranes), similar to our first patient.4 The individuals homozygous for GBA1 pathogenic variant c.1448T>C (p.Leu483Pro) (earlier known as L444P) present with severe disease and often with neurologic complications such as earlier onset and worsening of Parkinson’s disease.2 In the Indian spectrum, c.1448T>C is reportedly the most recurrent mutation, followed by D409H in patients of GD.5 The homozygosity for the complex allele L444P alongside E326 K mutation (c.1093G>A, exon 8) or heterozygosity for another allele such as P182L can be associated with severe PNL forms.6
| Type 1 | Type 2 | Type 3 | |
|---|---|---|---|
| Age of onset | 10–20 years of age | Infant to early childhood (3–6 months of age) | Childhood/adolescence |
| Cutaneous manifestations | Diffuse pigmentation/tanning/yellow-bronze pigmentation | Collodion baby/ichthyosis | Uncommon |
| Central nervous system involvement | Absent | Present | Present |
| Bone involvement | Present | Absent | Present |
| Hepatosplenomegaly | Present | Present | Present |
| Other features | Mucocutaneous bleeding (epistaxis/gingival bleeding/menorrhagia) may be present. |
Neck rigidity/opisthotonus, bulbar signs (swallowing difficulty), oculomotor paralysis (bilateral fixed strabismus). Perinatal lethal form: non-immune hydrops foetalis (dies in utero), pulmonary hypoplasia, pancytopenia, arthrogryposis, microcephaly, secondary progressive bulbar syndrome, dysmorphic facial features and severe neurological condition |
Gradual cognitive regression, calcification of cardiac valves/aorta |
| Disease prognosis | Varies from mild to severe | Worst prognosis | Slowly progressive, becomes severe later in childhood. |
| Histology changes | No epidermal abnormality | Dense hyperkeratosis, epidermal hyperplasia, inflammatory infiltrate consisting of lymphocytes and histiocytes – localised to the superficial epidermis | No epidermal abnormality |
| Stratum corneum – glucosylceramide to ceramide ratio | No change from normal | Increased | No change from normal |
| Laboratory investigations | |||
| 1) Angiotensin-converting enzyme | Increased in plasma of affected patients | ||
| 2) Beta-glucocerebrosidase activity assay | Direct assessment of enzyme responsible for disease | ||
| 3) Bone marrow aspirate | Visualisation of Gaucher cells in bone marrow | ||
| 4) Haematological tests | Anaemia and thrombocytopenia – hallmark features | ||
| 5) Liver function test | Hepatic dysfunction related to liver infiltration is common | ||
| Imaging- | |||
| 1) Ultrasonography abdomen | Hepatosplenomegaly, splenic infarcts | ||
| 2) X-ray findings | Lytic lesions, osteopenia, osteonecrosis, medullary infarcts, Erlenmeyer-flask deformity, endosteal scalloping. | ||
| 3) MRI Brain | Cerebral atrophy, dural thickening in cases with neuronal involvement. | ||
The PNL phenotypic type of GD is sporadic and has only been mentioned in a single case report by Aggarwal et al. from India, who found a heterozygous mutation of the RecNcil allele (missense mutation of three genes, namely, L444P, A456P and V460V) in both the parents of the foetus with gross intrauterine anomalies.7
This case report emphasises that severe ichthyosis in association with systemic complications may indicate GD. Genetic testing helps in the identification of this metabolic disorder and clears up the path for appropriate genetic counselling for future pregnancies.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Reference
- Global incidence and prevalence of Gaucher disease: A targeted literature review. J Clin Med. 2022;12:85.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Gaucher disease. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews. Seattle (WA): University of Washington, Seattle; p. :1993–2024. Hughes DA, Pastores GM. Gaucher disease. In: Adam MP, Feldman J, Mirzaa GM, et al, GeneReviews®. Seattle (WA). University of Washington, Seattle. 2000:1993-2024
- [Google Scholar]
- Perinatal-lethal Gaucher disease. Am J Med Genet A. 2003;120A:338-44.
- [CrossRef] [PubMed] [Google Scholar]
- Type 2 Gaucher disease: Phenotypic variation and genotypic heterogeneity. Blood Cells Mol Dis. 2011;46:75-84.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The mutation spectrum in Indian patients with Gaucher disease. Genome Biol. 2011;12:P26.
- [CrossRef] [Google Scholar]
- Perinatal lethal phenotype with generalized ichthyosis in a type 2 Gaucher disease patient with the [L444P;E326K]/P182L genotype: Effect of the E326K change in neonatal and classic forms of the disease. Blood Cells Mol Dis. 2005;35:253-8.
- [CrossRef] [PubMed] [Google Scholar]
- Molecular studies on parents after autopsy identify recombinant GBA gene in a case of Gaucher disease with ichthyosis phenotype. Am J Med Genet A. 2015;67:2858-60.
- [CrossRef] [Google Scholar]




