Translate this page into:
Genetic study in a suspected case of Schöpf-Schulz-Passarge syndrome
Correspondence Address:
Alejandro Vilas-Sueiro
Department of Dermatology, Avenida Residencia SN, 15405 Ferrol (A Coruña)
Spain
| How to cite this article: Vilas-Sueiro A, Monteagudo B, Gonz�lez-Vilas D, Varela-Veiga A, De las Heras C. Genetic study in a suspected case of Schöpf-Schulz-Passarge syndrome. Indian J Dermatol Venereol Leprol 2015;81:408-410 |
Sir,
To the present day, over 200 types of ectodermal dysplasias have been described; it is therefore a challenge to classify and diagnose these patients. We present a case of Schöpf-Schulz-Passarge syndrome in which genetic study demonstrated two homozygous missense mutations in WNT10A gene.
A 70-year-old male had no secondary dentition since childhood [Figure - 1]a. Since many years, he had a history of numerous yellow cysts around the eyelids [Figure - 1]b, several little papules on both helices [Figure - 1]c, generalized nail dystrophy resembling a pterygium [Figure - 2]a and palmo-plantar keratoderma [Figure - 2]b and c. Two of the his brothers and one of his sisters had identical complaints and a disease course similar to his.
 |
| Figure 1: (a) Hypodontia of the permanent teeth. (b) Multiple hydrocystomas around the eyelids. (c) Tumoral lesions in the left ear |
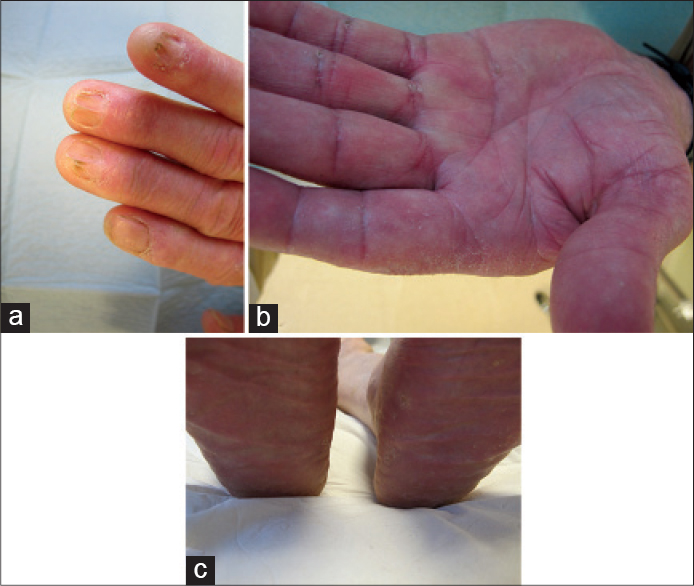 |
| Figure 2: (a) Onychodystrophy with severe pterygium. (b) Diffuse palmar hyperkeratosis. (c) Plantar keratoderma with desquamation |
Punch biopsy from a helix lesion revealed a dilated primary follicle lined by infundibular, stratified squamous epithelium which may have been connected to the epidermis, compatible with trichofolliculoma.
Following informed consent, genomic DNA was extracted from peripheral blood. Polymerase chain reaction was performed using four primer pairs spanning the coding exons and flanking introns of WNT10A. Direct sequencing demonstrated the presence of two homozygous single nucleotide transversion c. 682T>A and c.831G>T (Gen Bank NM_025216.2) which convert phenylalanine into isoleucine and tryptophan into cysteine. These missense mutations are designated p.Phe228Ile and p.Trp277Cys. These clinical findings and genetic results were consistent with a diagnosis of Schöpf-Schulz-Passarge syndrome. Our patient represents the second case of this syndrome having the missense mutation p.Trp277Cys. [1]
This syndrome is a rare genodermatosis with autosomal recessive transmission caused by mutations in the WNT10A gene described by Schöpf et al. in 1971. The gene encodes wingless-type MMTV integration site family member 10A, a small secreted signaling molecule that is a key in multiple developmental processes such as hair follicle morphogenesis and skin and tooth embryogenesis. In adult tissues, WNT10A inhibits the β-catenin degradation complex and is involved in tooth and hair follicle morphogenesis. [2],[3] Individuals who are heterozygous for WNT10A mutations may show ectodermal anomalies as dental, hair or nail defects. It has been calculated that approximately 50% of all individuals who are heterozygous for a missense mutation will show some feature of ectodermal abnormalities. [3]
Clinically, Schöpf-Schulz-Passarge syndrome is characterized by multiple eyelid apocrine hidrocystomas, hypodontia, hypotrichosis, nail dystrophy and palmo-plantar keratoderma. However, the phenotypic spectrum is variable, ranging from full-blown phenotypes to milder forms limited to isolated palpebral and tooth involvement. Clinical diagnosis of this syndrome was not established until 30-50 years ago, underscoring the lack of specificity of the initial ectodermal defects. Patients may show concomitant presence of eccrine syringofibroadenomas and other adnexal skin tumors. However, the risk of visceral malignancies is not increased. [3],[4],[5]
Odonto-onycho-dermal dysplasia and Schöpf-Schulz-Passarge syndrome are considered variable expressions of the same spectrum of WNT10A mutations. The most frequently observed nonsense mutation is p.Cys107X and the missense mutation is p.Phe228Ile (as in our case). The difference between these two syndromes is the onset of eyelid cysts, which provides a useful clue to the underlying diagnosis of Schöpf-Schulz-Passarge syndrome, although these should not be considered pathognomonic. In odonto-onycho-dermal dysplasia this feature is lacking, but it is frequent in the presence of a smooth tongue or erythematous facial lesions. [3],[4] Two further conditions, Bazex-Dupré-Christol and Rombo syndromes show features of ectodermal dysplasia with adnexal tumors. However, the absence of syringofibroadenomas and mutations in the WNT10A gene facilitate differential diagnosis. [5]
These patients should be assessed by a multidisciplinary team and should be followed regularly regarding the possible risk for the development of skin neoplasms.
| 1. |
Van den Boogaard MJ, Créton M, Bronkhorst Y, van der Hout A, Hennekam E, Lindhout D, et al. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet 2012;49:327-31.
[Google Scholar]
|
| 2. |
Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 2004;20:781-810.
[Google Scholar]
|
| 3. |
Petrof G, Fong K, Lai-Cheong JE, Cockayne SE, McGrath JA. Schöpf-Schulz-Passarge syndrome resulting from a homozygous nonsense mutation, p.Cys107X, in WNT10A. Australas J Dermatol 2011;52:224-6.
[Google Scholar]
|
| 4. |
Verplancke P, Driessen L, Wynants P, Naeyaert JM. The Schöpf-Schulz-Passarge syndrome. Dermatology 1998;196:463-6
[Google Scholar]
|
| 5. |
Castori M, Ruggieri S, Giannetti L, Annessi G, Zambruno G. Schöpf-Schulz-Passarge syndrome: Further delineation of the phenotype and genetic considerations. Acta Derm Venereol 2008;88:607-12.
[Google Scholar]
|
Fulltext Views
3,298
PDF downloads
1,588





![[Figure - 1]](#fig_ijdvl_2015_81_4_408_158657_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_4_408_158657_f2.jpg){kind=link}