
Translate this page into:
Genetics for dermatologists. Part 1: Fundamental concepts of structure, function, and clinical application

Corresponding author: Dr. Divya Gupta, Department of Dermatology, Dr BR Ambedkar Medical College, Bengaluru, Karnataka, India. divya_gupta@ymail.com
-
Received: ,
Accepted: ,
How to cite this article: Gupta D, Jose TG, Vishwanathan GB. Genetics for dermatologists. Part 1: Fundamental concepts of structure, function, and clinical application. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_243_2025
Abstract
Dermatology encompasses a broad range of conditions, ranging from single-gene disorders to common complex disorders characterised by polygenic risk factors like psoriasis and atopic dermatitis. For clinicians, understanding the genetic basis of these conditions is essential for accurate diagnosis, effective management, and appropriate genetic counselling. This article aims to revisit fundamental genetic concepts, offering a foundation to a better understanding of their application in clinical practice. It introduces the fundamental genetic terminology relevant for dermatologists, along with types of mutations, inheritance patterns, key elements of pedigree charts, and major online genetic databases, which serve as valuable resources for interpreting test results and identifying disease-associated variants. Additionally, it briefly explores the basics of genetic epidemiology, including linkage analysis, and association studies.
Keywords
Chromosome
gene
genetic epidemiology
patterns of inheritance
pedigree chart
mutation

Introduction
With the increasing availability of genetic testing in clinical dermatology, there is a growing need for dermatologists to understand core genetic concepts. This review provides a practical overview of fundamental principles, including gene structure and function, types of mutations, inheritance patterns, and interpretation of pedigree charts. It also introduces key online genetic databases and basic genetic epidemiology relevant to dermatological disorders. The aim is to equip clinicians with the essential principles needed to interpret genetic information in everyday practice.
Structure and function of genes
Chromosomes, DNA, and genes
Human cells contain 46 chromosomes, arranged in 23 pairs—22 pairs of autosomes (non-sex chromosomes) and one pair of sex chromosomes (XX in females and XY in males). A chromosome is a tightly coiled strand of DNA combined with proteins called histones [Figure 1a]. Genes, the functional units of hereditary characteristics, encode specific proteins but make up only a small portion of DNA; rest is the intergenic portion that has no known function. DNA consists of two strands forming a double helix, each with a sugar-phosphate backbone linked by covalent bonds. Nitrogenous bases—adenine (A), thymine (T), cytosine (C), and guanine (G)—pair through hydrogen bonds, A pairing with T and C with G. The strands run in opposite directions (antiparallel), ensuring complementarity and polarity, with one end marked 5’ (phosphate) and the other 3’ (hydroxyl). During DNA replication, these strands separate, and new complementary strands are synthesised based on base-pairing rules. This ensures accurate replication of genetic information. Important terms related to the basics of genetic architecture have been described in Table 1.1
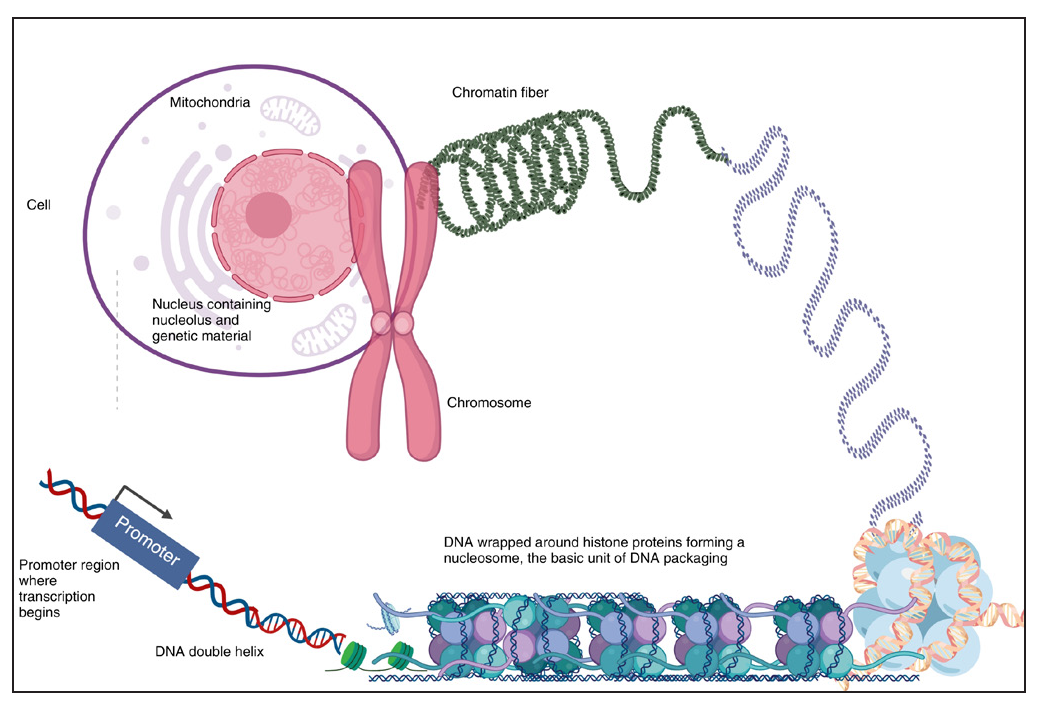
- Figure of human cell depicting chromosomes and DNA.
| Term | Definition |
|---|---|
| Genome | The complete set of genetic material (DNA) in an organism, including all coding and non-coding regions. |
| Exome | The part of the genome that consists of all exons, representing the coding regions for proteins. |
| Locus | The specific location of a gene or DNA sequence on a chromosome. |
| Allele | A different form or version of a gene at a particular locus. |
| Gene | A segment of DNA that contains instructions for making a specific protein or performing a particular function, influencing traits and biological processes. Every gene is made up of exons and introns |
| Exon | A coding section of a gene that is used to make proteins. |
| Intron | A non-coding section of a gene that is removed during RNA processing and does not code for proteins. |
| Genotype | The genetic makeup of an individual, specifically the combination of alleles they carry. This is the internally coded, inheritable information or the basic genetic constitution that a person receives from their parents. Thus, it is the blueprint of the structure and function for an individual. |
| Phenotype | The observable traits or characteristics of an individual resulting from both genetic and environmental factors. |
| Genotyping | Testing to identify an individual’s genetic variants at specific locations. |
| Phenotyping | The process of assessing and documenting an individual’s observable traits or clinical features often used to link physical characteristics with genetic findings in diagnosis |
| Heterozygote | An individual with two different alleles for a particular gene, one from each parent (e.g., Aa). |
| Homozygote | An individual with two identical alleles for a particular gene, either both dominant (AA) or both recessive (aa). |
| Hemizygote | An individual with only one copy of a gene in a diploid cell often due to the gene being on the X chromosome in males (e.g., X-linked genes in males). |
| Autosomal dominant | A trait or disorder that appears when only one copy of a mutated gene on a non-sex (autosomal) chromosome is present. E.g. Neurofibromatosis 1 |
| Autosomal recessive | A trait or disorder that appears only when two copies of a mutated gene on a non-sex (autosomal) chromosome are present. E.g., Recessive dystrophic epidermolysis bullosa |
| X-linked dominant | A trait or disorder caused by a mutation on the X chromosome, expressed even if only one mutated gene is present (e.g., Incontinentia pigmenti). Affects both males and females, though often more severe lethal in males. |
| X-linked recessive | A trait or disorder caused by a mutation on the X chromosome, usually expressed only when both X chromosomes carry the mutation in females or the single X in males. E.g. Wiskott Aldrich syndrome |
| Monogenic inheritance | A trait or disorder caused by a mutation in a single gene. E.g., Neurofibromatosis 1 is caused by a mutation gene neurofibromin |
| Polygenic/Multifactorial inheritance | Multiple genes, along with environmental factors, influence a trait, with each contributing a small effect to the overall phenotype. Examples include skin colour, height, and susceptibility to conditions like psoriasis, atopic dermatitis, systemic sclerosis, etc. |
| Mitochondrial inheritance | Each mitochondrion contains many copies of a circular double-stranded DNA molecule. Genes in mitochondrial DNA are inherited exclusively from the mother, as sperm do not contribute mitochondria to the embryo - and thus show maternal inheritance. E.g., Leber’s hereditary optic neuropathy. Mutations in mitochondrial DNA lead to disruptions in energy metabolism. |
| Anticipation | A pattern where a genetic disorder appears at an earlier age or with increased severity in successive generations, often seen in disorders with repeat expansions like Huntington’s disease. The genes associated with these conditions contain a series of repeated triplets of DNA bases (e.g., CAGCAGCAG...). An abnormal increase in the number of these repeats during DNA replication leads to unstable sequences, with offspring inheriting longer repeats than their parents. Anticipation occurs because the larger the repeat size, the more severe the disorder and the greater is the chance of further expansion in the next generation. |
| Imprinting | An epigenetic phenomenon where the expression of a gene depends on whether it is inherited from the mother or the father. In imprinted genes, only one parental allele is active, while the other is silenced through DNA methylation or histone modification. Disruption of imprinting can lead to disorders such as Prader-Willi syndrome (paternal allele loss) and Angelman syndrome (maternal allele loss). |
| Uniparental disomy | A situation where both copies of a chromosome are inherited from one parent instead of one from each, which can lead to genetic disorders if imprinted genes are involved. |
| Clinical heterogeneity | Mutations in the same gene causing different clinical conditions. E.g. Mutations in the XPD gene can cause xeroderma pigmentosum, Cockayne syndrome or trichothiodystrophy |
| Genetic heterogeneity | Mutations in different genes causing the same clinical condition E.g.- mutations in 9 genes can cause ARCI |
| Proband/Index case | The first individual in a family identified with a genetic condition, who is used as a reference for family studies. |
| Carrier | A person who has one normal allele and one mutated allele for a recessive genetic condition, typically without showing symptoms but capable of passing the mutation to offspring. |
ARCI: autosomal recessive congenital ichthyosis
Transmission of genetic information
Genes are the basic units of heredity, encoding information to produce functional proteins. They consist of coding regions (exons) and noncoding regions (introns). The coding sequence contains triplets of nucleotide bases (codons), each representing an amino acid [Figure 1b]. This genetic code is nearly universal. Since there are 64 possible codons but only 20 amino acids, some codons encode the same amino acid. This phenomenon is called redundancy or degeneracy of the genetic code.

- Triplet codon representing various amino acids. (A: adenine, T: thymine, C:cytosine, G: guanine, U: uracil)
Protein production involves two steps: transcription, where DNA is used as a template to generate messenger RNA (mRNA), and translation, where mRNA directs the assembly of amino acids into a functional polypeptide. This flow of information from DNA to RNA to protein is called the central dogma of molecular biology [Figure 1c].
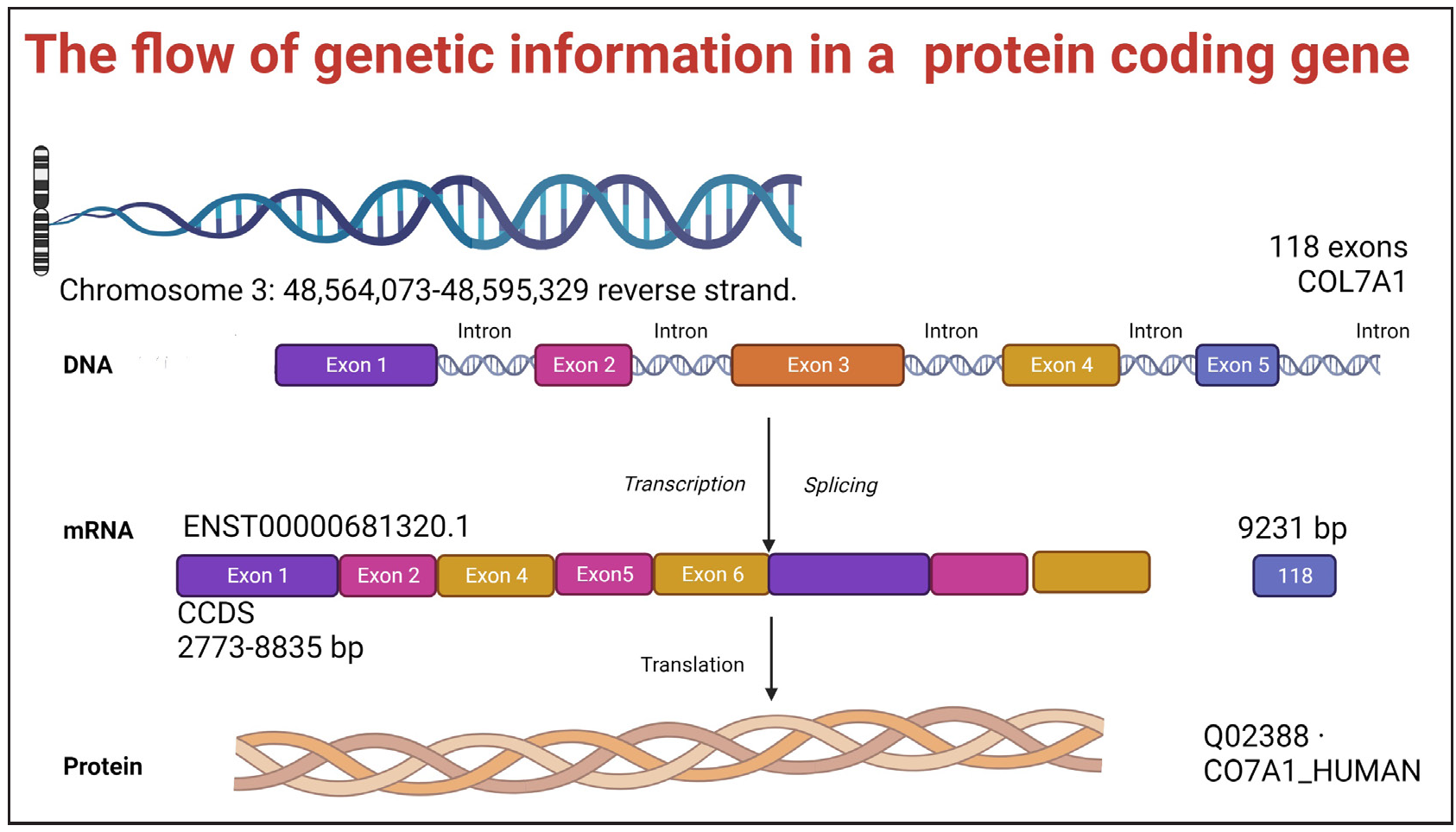
- Flow of genetic information in the COL7A1 gene located on the reverse strand of chromosome 3 (position 48,564,073–48,595,329). The DNA segment contains exons (coloured blocks) and introns (thin lines) spanning 118 exons. During transcription, the entire gene is copied into pre-mRNA. Introns are removed during splicing, generating mature mRNA (transcript ENST00000681320.1, 9231 bp). Translation of this mRNA produces the collagen VII protein (Q02388· CO7A1_HUMAN), a crucial structural component of anchoring fibrils in the skin.
Transcription overview
During transcription, one DNA strand serves as a template to produce a complementary mRNA molecule, synthesised in the 5’ to 3’ direction by RNA polymerase using RNA precursors (ATP, CTP, GTP, UTP). In mRNA, uracil replaces thymine. Gene expression is highly regulated via promoter sequences (e.g., TATA, GC, CAAT boxes) near the gene and distant regions called enhancers that increase transcription efficiency.2,3
Post-transcriptional processing
The primary RNA transcript includes both exons and introns. To create mature mRNA, introns are precisely removed, and exons are joined through mRNA splicing. Errors in splicing due to splice-site mutations can potentially delete exons, leading to functional defects.
Translation overview
After mRNA is synthesised in the nucleus, it moves to the cytoplasm and associates with ribosomes, which facilitate protein synthesis. Ribosomes work with mRNA and tRNA during translation. Each tRNA carries a specific amino acid and has an anticodon that pairs with the corresponding mRNA codon. Translation begins at the AUG start codon near the 5’ end of mRNA, establishing the reading frame. Methionine is the first amino acid to be added, followed by others, as tRNAs sequentially bind codons and form peptide bonds. This cycle continues until a stop codon (UAA, UGA, or UAG) signals termination, releasing the complete polypeptide.
Post-translational modifications
Once synthesised, many proteins undergo modifications to achieve proper structure and function. These include cleavage (e.g., proinsulin to insulin), the addition of groups like carbohydrates (glycosylation) or lipids (palmitoylation), and chemical alterations such as phosphorylation or methylation. These modifications are essential for protein activity and targeting them to their final cellular destinations, such as the cell membrane.4
An overview of transcription and translation is shown in Figure 1c.
Patterns of genetic transmission
Every individual inherits two copies of genes—one from each parent—except for those located on the sex chromosomes. These genes that occupy the same location on paired chromosomes with different forms are known as alleles. The combination of alleles at a specific location (locus) forms the genotype, which determines the observable physical characteristic or trait called the phenotype. Genetic transmission can be classified as either Mendelian or non-Mendelian, depending on the pattern of inheritance.5
Mendel’s laws of inheritance
Mendel’s laws of inheritance form the basis of classical genetics and describe how traits are passed from parents to offspring. His first law, the Law of Segregation, states that every individual has two alleles for a gene, which separate during gamete formation, ensuring that each gamete receives only one allele. The second law, the Law of Independent Assortment, explains that different genes are inherited independently of one another, provided they are on different chromosomes. Mendel’s third law, the Law of Dominance, states that some alleles are dominant and can mask the effect of recessive alleles.6
Mendelian inheritance patterns
These can broadly be described as 1) autosomal dominant, 2) autosomal recessive, 3) X-linked dominant, and 4) X-linked recessive [Table 1, Figure 2].
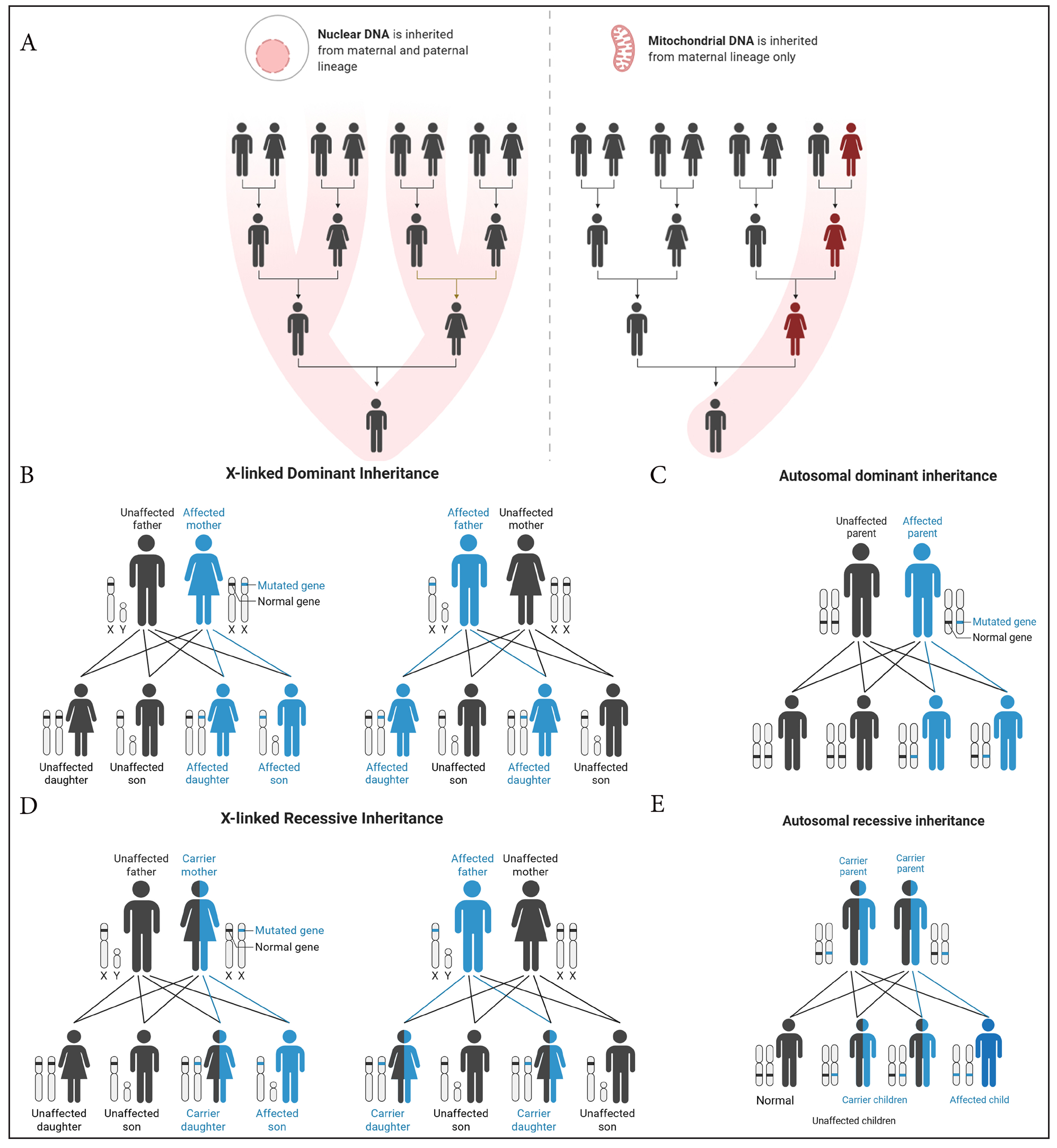
- Patterns of genetic inheritance.
Autosomal Dominant: A condition that manifests when a person inherits just one mutated gene from either parent. Affected individuals have a 50% chance of passing it to their children (e.g., tuberous sclerosis, Darier’s disease).
Autosomal Recessive: A disorder that occurs only when both copies of a gene (one from each parent) are mutated. Carriers (who have one normal and one mutated gene) do not show symptoms (e.g., congenital ichthyosis, epidermolysis bullosa simplex).
X-Linked Dominant: A mutation in a gene on the X chromosome that causes disease even if only one copy is altered, while affected females have a 50% chance of passing it to children (e.g., incontinentia pigmenti).
X-Linked Recessive: A condition caused by mutations in genes on the X chromosome, primarily affecting males, as they have only one X chromosome. Females are usually carriers unless both X chromosomes are affected (e.g., chronic granulomatous disease). Affected males pass it to all daughters but not sons.
Non-mendelian inheritance patterns
Non-Mendelian inheritance refers to genetic patterns that do not follow Mendel’s laws. Non-Mendelian patterns relevant to dermatology have been discussed below and the remaining have been described in Table 1.7
Co-dominance: Both alleles in a pair are fully expressed in the phenotype, such as in blood group type AB, where both A and B antigens are present.
Incomplete dominance: Phenotype is an intermediate of the two alleles, thus showing a blend of traits. E.g., the genes that produce the melanin (pigment) for either dark or light skin, wavy hair, etc.
Pseudo dominance: When a recessive disorder appears to follow a dominant inheritance pattern, often because the affected individual has a child who is a carrier, making the trait appear in consecutive generations.
Paradominance: A rare inheritance pattern where a mutation only causes symptoms if it occurs in a mosaic form rather than being present in every cell. Happle proposed this to explain why a mosaic phenotype that usually occurs sporadically, such as Becker naevus, may rarely affect several family members. It has also been used to explain Klippel–Trenaunay syndrome, sebaceous naevus, and cutis marmorata telangiectatica congenita.8
Epitasis: The effect of one gene is influenced by other gene(s). Essentially, one gene can “mask” or modify the effect of another gene. E.g., in albinism, caused by a mutation in the tyrosinase gene, a person will have pale skin, regardless of the skin colour genes that were inherited. Similarly, In a person with a gene for baldness (which causes hair loss), the effect of hair colour genes becomes irrelevant—because without hair, hair colour cannot be expressed. The baldness gene masks the expression of hair colour (i.e. one gene’s effect overrides another).
Epigenetics: The study of heritable changes in gene expression that do not involve alterations in the DNA sequence itself. Epigenetic mechanisms, such as DNA methylation and histone modification, affect health and disease by influencing how genes are turned on or off without changing the underlying genetic code. Epigenetic factors have been proposed to play a role in various inflammatory skin conditions like psoriasis, systemic lupus erythematosus, etc.
Expression (or expressivity): Refers to the degree or severity of the phenotype expressed by individuals with a particular genetic variant. When the same genetic mutation causes a range of different phenotypic manifestations, from mild to severe, it is said to have variable expression. For example, in a disorder like neurofibromatosis type 1 (NF-1), individuals may exhibit a wide variety of symptoms, from café-au-lait spots to severe neurological complications.9,10
Penetrance: Refers to the proportion of individuals carrying a particular genetic variant (mutation),expressing the associated phenotype (trait or condition) over time. It is usually expressed as a percentage. If some individuals with the genetic variant do not express the associated trait, it is considered incomplete penetrance. For example, if only 70% of individuals with a specific mutation show symptoms of the disease, the penetrance is 70%.11
Mosaicism: It is a genetic phenomenon where an individual has two or more genetically distinct cell populations within their body, arising from mutations that occur after fertilisation. This means that only some cells carry the mutation, while others do not, leading to a “mosaic” pattern of affected and unaffected cells [Figure 3]. In dermatology, this results in altered pigmentation or texture in localised areas corresponding to mutations present in those skin cells. E.g., in verrucous epidermal naevus, a post-zygotic keratin gene mutation causes linear hyperkeratotic, verrucous plaques that follow Blaschko’s lines. Only these specific skin regions carry the mutation, giving a “mosaic” appearance. Similarly, in segmental NF1, postzygotic somatic NF1 gene mutations affect only a subset of the body’s cells in contrast to the classic generalised form, where a germline NF1 mutation is present in all diploid cells. In the segmental variant, the typical NF1 features like café-au-lait macules (CALMs), skinfold freckling, and neurofibromas are confined to a specific body segment. In such cases, genetic testing will identify a somatic NF1 mutation in affected skin but not in blood, making diagnosis challenging.12
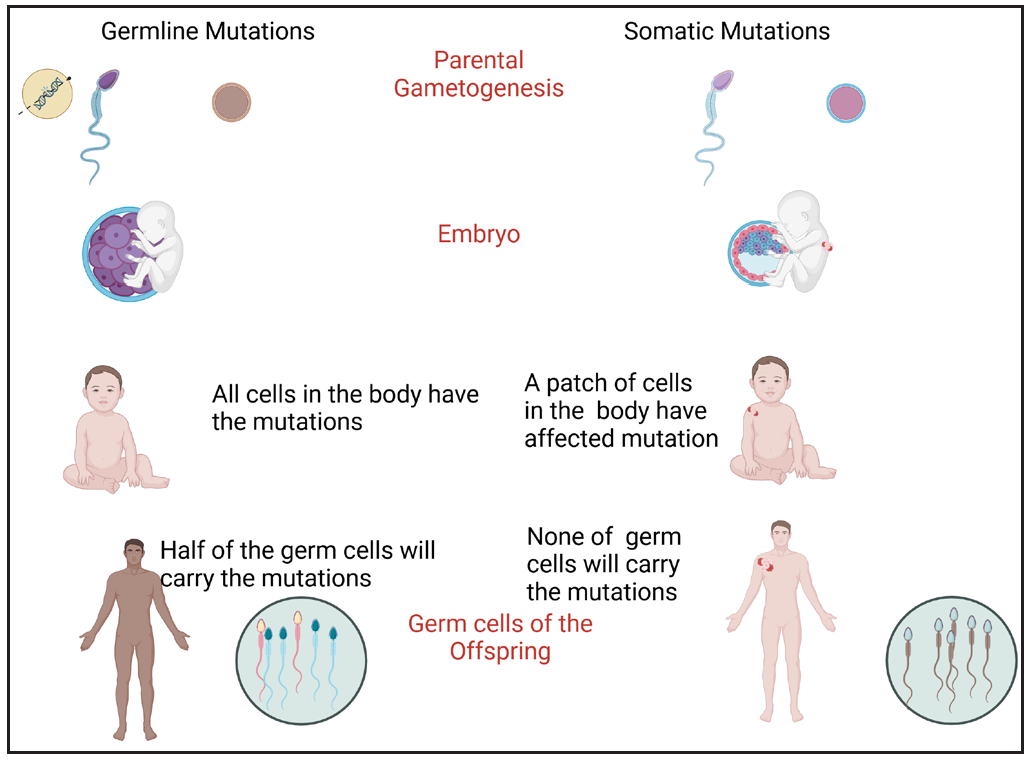
- Principle of germline mutation vs somatic mutation (the latter giving rise to mosaicism).
Some authors consider incomplete dominance, codominance, multiple alleles, and sex-linked traits as an extension of the Mendelian laws since there are alterations in the phenotypic ratios, but the genotypic ratios remain the same.5
For interested readers, the different types of mosaicism and additional genetic terminologies have been covered in Supplementary Material 1.
Gene mutations (variation/variant)
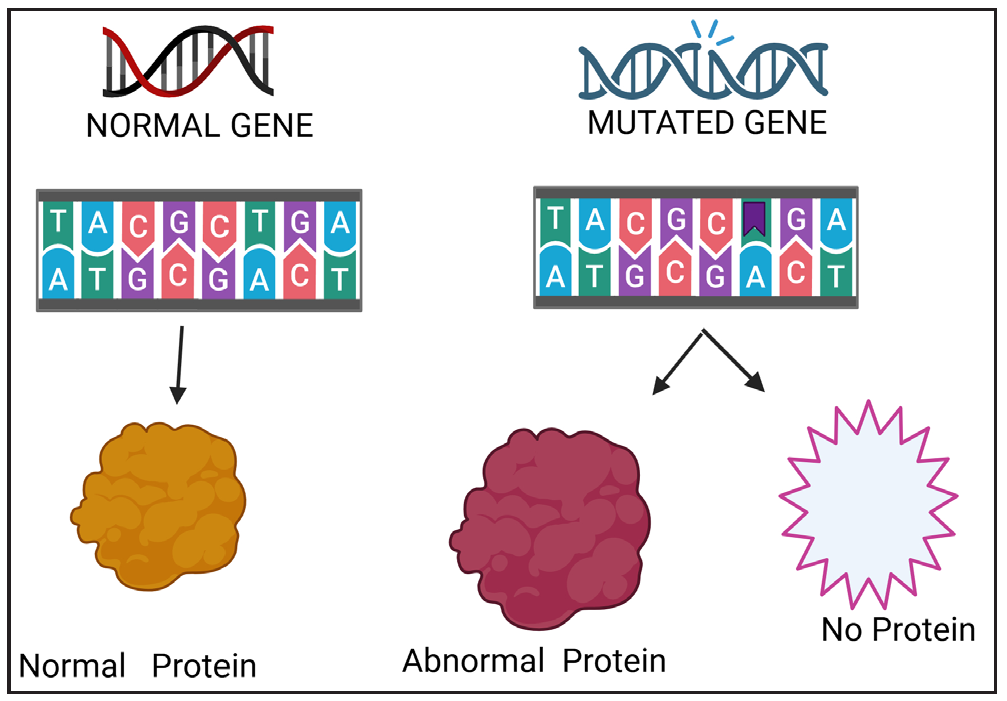
A protein’s properties are determined by its sequence of amino acids, which is dictated by the base sequence of the gene that encodes it. Changes in this DNA sequence, known as mutations, lead to a change in the structure and/or function of the resultant protein. The frequency is less than 1% of the population.3,13,14
The American College of Medical Genetics and Genomics (ACMG) advocates replacing the term “mutation” with “variant” or “variation” to reduce the misconception that all genetic changes are harmful. The term “variant” allows for a more precise classification based on clinical significance, distinguishing between benign, likely benign, uncertain significance, likely pathogenic, and pathogenic changes in the genome.
The typical or most common genetic variant of a gene found in a natural population, is the wild type mutation, which is generally associated with the standard or “normal” phenotype. Wild type is often used as a reference to compare the mutated forms of a gene that may cause disease. Types of mutations have been described in Table 2, Figures 4a-e.15,16
| Mutational Cause | Explanation | Example |
| Spontaneous (de novo) | Due to replication errors evaded from proof-reading mechanisms at the time of meiosis. | Autosomal dominant conditions where variant is reported in index case but not found in parents. |
| Mutagens induced | Due to the reaction with parental DNA inducing structural change that affects base pairing. | XP patients on exposure to UV light. |
| Mutational location | ||
| Somatic mutation | Acquired mutations in the somatic cells that cannot be passed on to offspring | Mosaic conditions like hypomelanosis of Ito, some types of epidermal nevi |
| Germline mutation | Mutations in the reproductive cells that are passed on to offspring. | Most single gene disorders, e.g., xeroderma pigmentosum, neurofibromatosis, tuberous sclerosis, epidermolysis bullosa |
| Mutational effects | ||
| Gain of function (GOF) | Mutations that give to rise to abnormal gene products which function with newer patterns of expression and molecular function. Mostly dominant or semi-dominant in nature. | Over 90% cases of cutaneous mastocytosis harbour a GOF mutation in the KIT receptor tyrosine kinase, which results in enhanced survival and growth of neoplastic mast cells. |
| Loss of function (LOF) |
Mutations result in altered gene products, which have reduction in the molecular function. Mostly recessive mutations. |
Recessive Dystrophic Epidermolysis Bullosa is due to LOF mutation in COL7A1, resulting in reduced or absent Collagen 7, which affects the integrity of dermo-epidermal junction and predisposes to increased skin fragility. |
| Mutational effects on genome* | ||
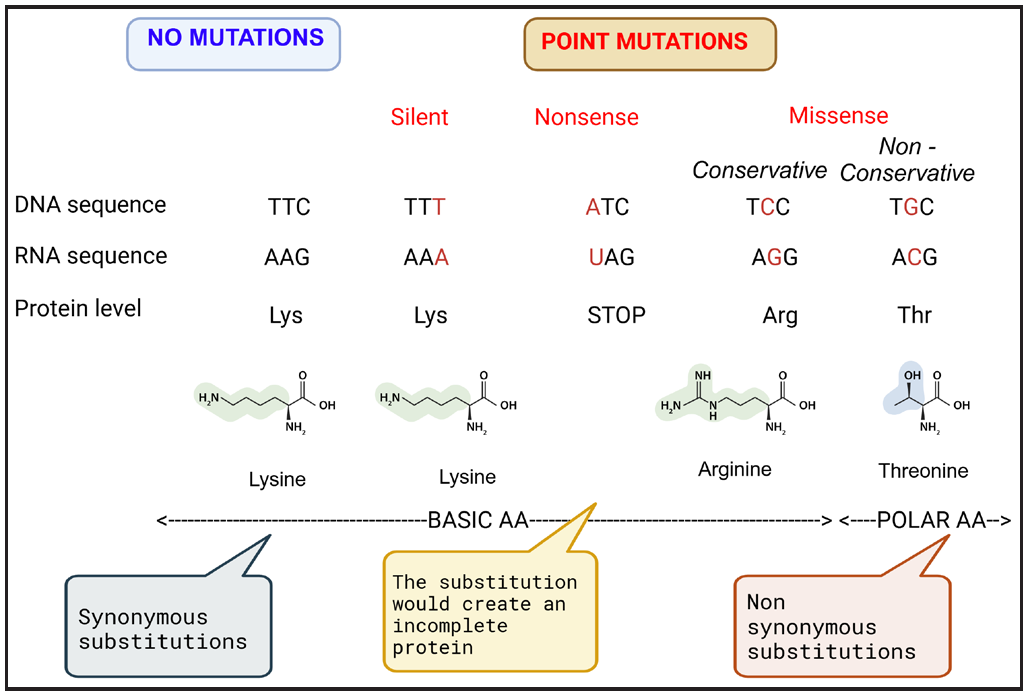
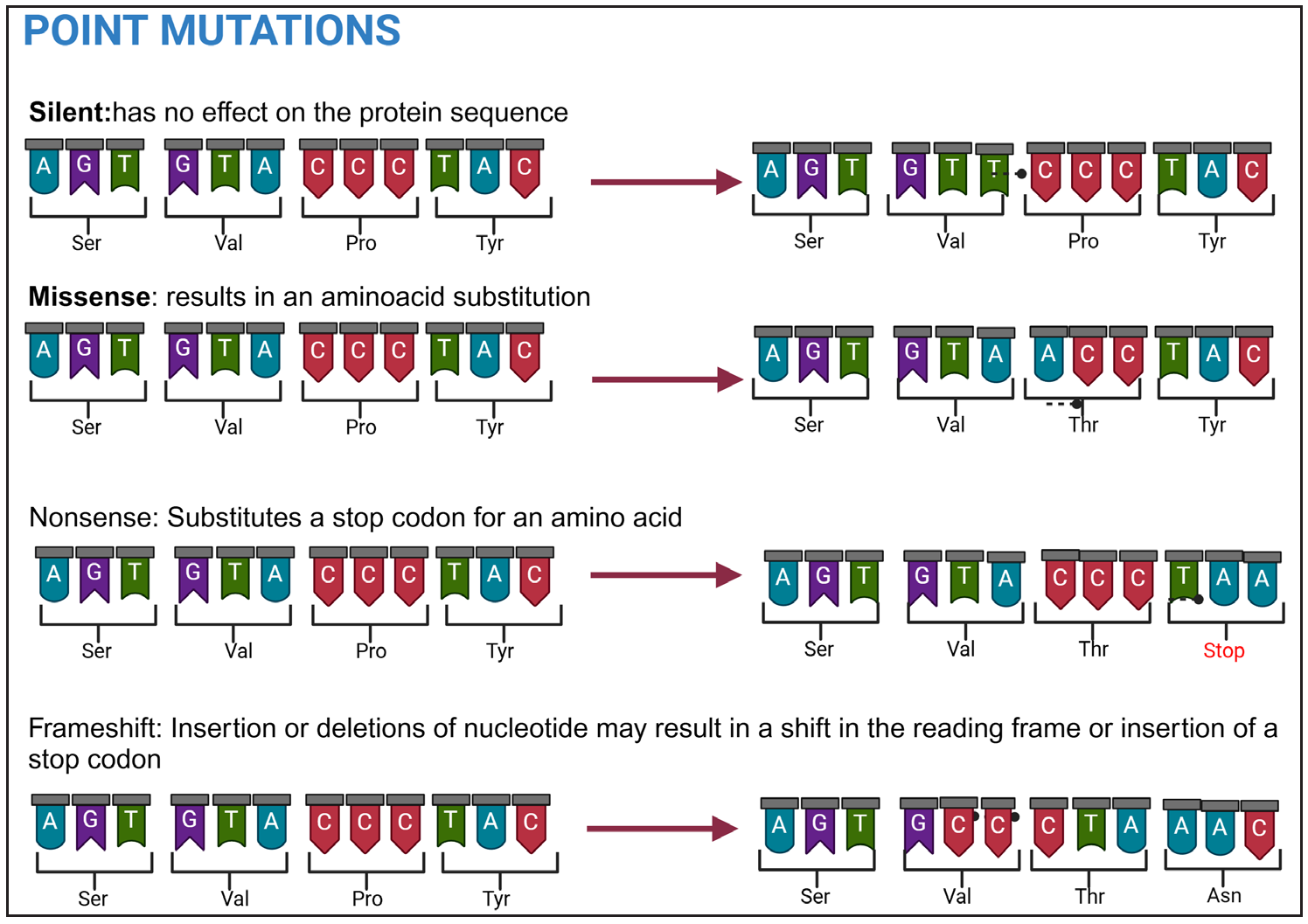
| Synonymous mutation (silent mutation/codon substitutions) | New codon encodes the same AA with no resultant effect on protein | Both UUU and UUC code for phenylalanine; substitution of last base (U with C) may not affect protein function |
| Non-Synonymous mutation |
Missense mutation, which results in a different AA or an AA substitution that may change the chemical properties of the protein. If the AA has similar chemical properties then the mutation is said to be conservative and has minimal functional impact. Significant changes can result in loss, gain, or alteration of function. |
|
| Nonsense/Stop mutation results in a stop codon (UAA, UAG, UGA) and hence termination of the protein translation. | ||
| Readthrough/Nonstop mutation results in a readthrough of the stop codon signal | ||
| Mutation types | ||
| Point mutation | ||
| Substitution | Transition | Interchange between purines or between pyrimidines. |
| Transversion | Change occurs between purines and pyrimidines | |
| Insertion(ins) | Small or large insertion | A new base pair (bp) gets inserted into the sequence. Depending upon the number of nucleotides it can be a large or small insertion. E.g., small in-frame insertions or deletions in KRT6A/6B can lead to Pachyonychia Congenita. |
| Insertion induced Frameshift # | The reading frame gets altered if the mutation results in the insertion of nucleotides, not in the multiples of three | |
| Deletion(del) | Small or large deletion | Base pair(s) get deleted from the sequence. It can range from the removal of large sections of DNA, such as entire genes, to the loss of a single base, resulting in small or large deletions |
| Deletion induced Frameshift # | The reading frame gets altered if the mutation results in the deletion of nucleotides are not in the multiples of three. | |
| Trinucleotide repeat expansion | Abnormal repeats of three-nucleotide sequences (e.g., CAG, GAA) within a gene, which disrupts normal gene function, leading to disorders that worsen across generations due to anticipation. E.g., Huntington’s disease, Friedrich’s ataxia | |
| Splice site mutations | Mutations in the splice donor or acceptor sequences can lead to exon skipping or the inclusion of some intron sequence in the mRNA | |
| Chromosomal mutations | ||
|
Structural variations Large alterations in the genome, typically affecting regions larger than 50 base pairs, that change the structure of chromosomes. |
Inversion (inv) | The chromosomal region is flipped and reinserted. |
| Deletion (del) | Absence of the chromosomal region. | |
| Duplication (dup) | The chromosomal region gets repeated | |
| Translocation | A chromosomal region breaks off and attaches to another chromosomal region. | |
| Copy number variations (CNVs) | DNA segment is amplified or deleted, resulting in more or fewer than the normal two copies of a gene. CNVs are typically larger than 1 kb, but newer methods detect variants as small as 50 bp. | |
| Indels | Indels are small insertions or deletions of bases in the DNA sequence, typically ranging from 1 to 50 base pairs. They can disrupt gene function if they occur within coding regions. |
* In-frame single nucleotide changes that do not result in frame-shift mutation. # Frameshift mutation disrupts the codon sequence, often producing an altered protein sequence with premature stop codons. This typically results in a truncated, nonfunctional protein that is degraded in the cell. DNA: Deoxyribonucleic acid, XP: Xeroderma pigmentosum, UV: Ultraviolet, GOF: Gain of function, LOF: Loss of function, KIT: Tyrosine kinase receptor, A: Adenine, T: Thymine, C: Cytosine, G: Guanine, U: Uracil, AA: Amino acid; KRT: Keratin gene, CNV: Copy number variants
- Consequence of a mutation.
- Types of point mutations.
- Types of point mutations.
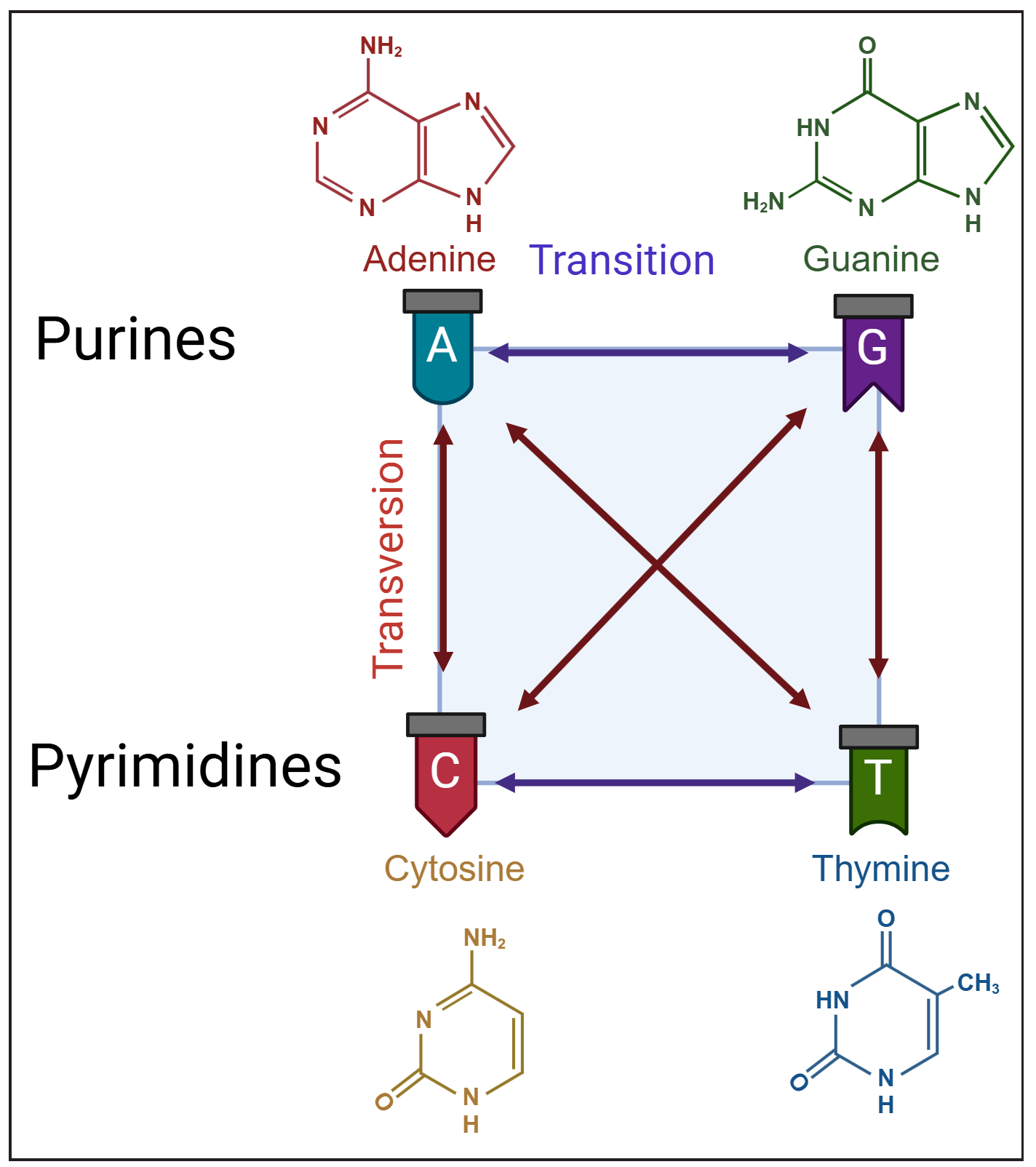
- Diagrammatic representation of nucleotide substitutions classified as transitions (purine-to-purine or pyrimidine-to-pyrimidine changes) and transversions (purine-to-pyrimidine or vice versa).
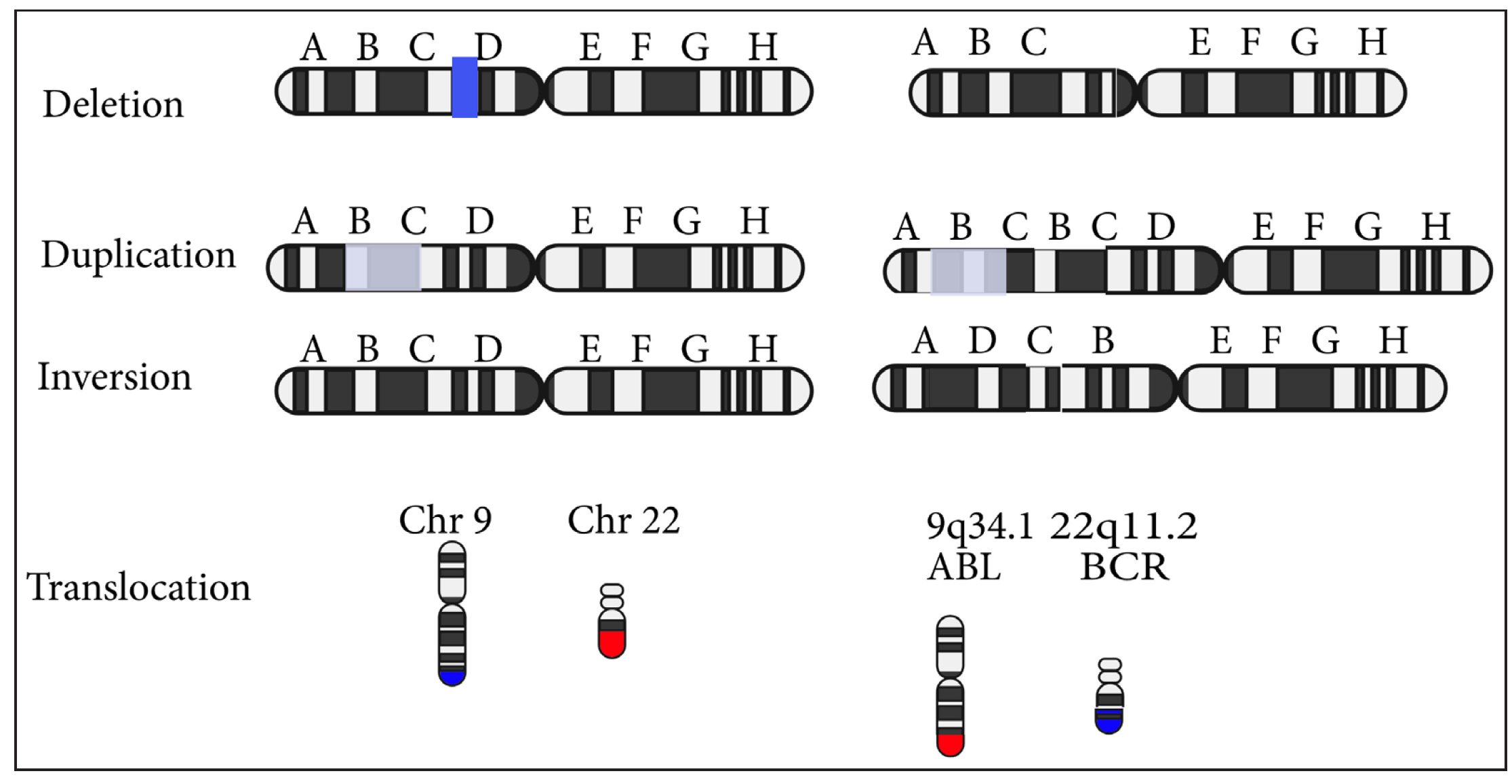
- Types of chromosomal mutations.
Pedigree charting and its components
A pedigree chart is a family tree diagram that tracks the inheritance of genetic traits or conditions across generations, helping identify inheritance patterns. A three-generation pedigree chart is necessary and helps the clinician to predict disease risk in subsequent generations, guides genetic testing, and aids genetic counselling, and medical decision-making. The important symbols of a pedigree chart have been depicted in Table 3 and additionally shown in Supplementary Material 2.17,18
| Basic symbols for individuals | Male | Square 
|
| Female | Circle 
|
|
| Unknown or unspecified sex (often used for foetuses) | Diamond 
|
|
| Marriage and mating | Mating | Horizontal line between individuals 
|
| Consanguineous mating | Double line between individuals 
|
|
| Children and generations | Connects parents to their children. | Vertical line down from mating line 
|
| Siblings | Arranged horizontally from the same vertical line, connected at the top 
|
|
| Affected or carrier status | Affected individual (shows the disease or trait) | Full shaded square or circle 
|
| Carrier of a recessive trait (typically unaffected by the condition but can pass it on) | Half-shaded square or circle 
|
|
| Carrier for X-linked recessive trait (usually used for female carriers) | Dot in the middle of the symbol 
|
|
| Deceased individuals | Indicates the person is deceased. | Diagonal line through symbol 
|
| Proband (First Affected individual studied) | Arrow pointing to symbol 
|
|
| Twins | Dizygotic (non-identical) twins |
|
| Monozygotic (identical) twins. |
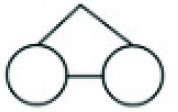
|
|
| Adoption | Adopted individuals, with brackets indicating they’re not biologically related | Square or circle with brackets 
|
Successive generations are numbered as I, II, III, and so on. Offsprings in each generation are numbered as I-1, I-2; II-1, II-2 and so on. Order of birth in a pedigree chart is from left to right.
Genetic epidemiology
Medical genetics relies heavily on the concept of genetic variation. These variations range from significant changes causing single-gene disorders like sickle cell anaemia to harmless differences, such as those determining hair or eye colour. Between these extremes lie variants that influence the risk of common conditions like hypertension or diabetes. The frequency of such variants differs across populations, contributing to certain groups’ predisposition to specific disorders. In this context, certain terms warrant a closer understanding:
A polymorphism is defined as any change in the DNA base sequence with a frequency of >1%. The most common type of polymorphism is a change in a single DNA base pair, called a single nucleotide polymorphism (SNP). SNPs occur roughly once every few hundred base pairs and are useful for gene mapping and studying the association of specific genes with complex traits. They can have functional implications when they occur within coding regions.
Other types of polymorphisms include changes in the number of repeated DNA sequences, either large blocks of tens or hundreds or short 2–4 base repeats. These are useful in genetic linkage studies.
Allele frequency is the proportion of a specific allele in a population, used to understand how common or rare a variant is. High allele frequency in the general population often suggests that a variant is likely benign, as it would be uncommon for a harmful (pathogenic) mutation to be so widespread without severe effects on health. Conversely, pathogenic variants—those associated with disease—are typically rare, as they often cause conditions that can affect survival, thereby limiting their spread in the population. Typically, an allele frequency less than 1% may suggest association with some disease.
The founder effect refers to a genetic phenomenon where a small group of individuals becomes isolated from a larger population, leading to endogamy and reduced genetic diversity. In this smaller group, certain rare genetic traits or mutations can become more common over generations. This results in specific genetic disorders appearing at higher frequencies than in the general population. For e.g., Ashkenazi Jews, Amish community, Bedouins, French Canadian population.
Genetic linkage and association
Mapping the thousands of genes in humans to their respective diseases is not easy. Two approaches can help us to study this relationship: 1) genetic linkage 2) association or candidate-gene approach1
Linkage studies rely on the fact that genes are arranged in a fixed linear order on chromosomes, consistent across individuals. Alleles of two closely linked genes tend to be inherited together unless a crossover occurs during meiosis between their loci. The frequency of these crossover events depends on the distance between the loci, allowing recombination rates to be used for genetic mapping. The human genome now contains a dense map of polymorphic loci with precisely known positions. These polymorphisms help identify loci that co-segregate with genetic disorders in families, enabling the mapping and eventual identification of disease-causing genes. This approach has led to the discovery of hundreds of new gene-disease connections over the last two decades.14
The linkage approach is effective for identifying single genes with a major influence on a disorder but is less suited for uncovering genes with smaller effects in multifactorial diseases. In such cases, genetic association analysis is used. This method often tests “candidate” polymorphisms—variants in genes believed to be involved in the disease process. Typically, a case-control study design is used to compare the frequency of specific alleles between affected and unaffected individuals. If an allele is more common in those with the disease, it is associated with the condition. However, there is a caveat here - this association may indicate that the allele itself contributes to the disease or that it is near another causative allele (and both are inherited together) - a phenomenon known as linkage disequilibrium.19
Association studies are constrained by the fact that individual alleles often have minor effects, necessitating large sample sizes for studies. Genetic contributions may differ across populations making it difficult to extrapolate the data to all individuals. The inherent disadvantage of the candidate-gene approach is that it limits the scope of discovery. Ideally, researchers should scan the entire genome for any allele associated with a disease, uncovering any unexpected links. However, the cost of sequencing such vast number of SNPs in large populations remains a significant barrier.20
A guide to navigating online genetic databases
These online databases maintain updated information about genetic skin diseases, including their genotype and phenotype. These are important sources of information for not just clinicians but also for patients. Some of them contain information on centres for genetic testing, counselling, and management of genetic conditions. Some genetic databases are essential for clinicians as they provide valuable information on genetic variants and associated diseases.21 These include:
OMIM (Online Mendelian Inheritance in Man)
Initially created by Dr. Victor A. McKusick as Mendelian Inheritance in Man, OMIM is now maintained at Johns Hopkins University and hosted by NCBI (www.ncbi.nlm.nih.gov/omim). It provides detailed summaries of genetic conditions and gene functions, with links to mutation databases, gene nomenclature, sequence data, protein structure, PubMed references, and patient resources. OMIM is searchable by specific clinical features, so it is also a valuable tool for differential diagnosis. A step-by-step guide on navigating OMIM has been described in Table 4.22–24
|
|
|
|
OMIM: Online mendelian inheritance in man, MIM: Mendelian inheritance in man, BRCA1: BReast CAncer gene 1, NCBI: National center for biotechnology information, gnomAD: Genome aggregation database, HGMD: Human gene mutation database
GeneReviews
GeneReviews is an internationally recognised clinical resource on inherited disorders, covering diagnosis, management, and genetic counselling. There are over 900 PubMed-indexed chapters written by subject experts, which are peer-reviewed before online publication. These are regularly updated every four to five years. Key terms within an article are hyperlinked to a glossary. The website also has resource materials for genetics professionals and patients. One can navigate disorder-wise to access peer-reviewed summaries.25
Orphanet
Established in 1997, in France by the INSERM (French National Institute for Health and Medical Research), Orphanet is now a consortium of 40 countries within Europe and globally, supported by grants from the European Commission. It provides information on rare diseases, the genes involved, orphan drugs, and a directory of patient organisations, professionals, institutions, expert centres, and medical labs providing diagnostic tests. One can use the search tool to find disease summaries, where links are available to access clinical trials, biobanks, registries, network of experts, and patient organisations pertaining to that disease.26
HGMD (Human gene mutation database)
A database of disease-causing mutations reported in literature and is maintained in Cardiff, UK. A less up-to-date public version is freely available to registered users from academic institutions/non-profit organisations. All other commercial users are required to purchase a license. Use search filters like gene, disease, or mutation type.27,28
ClinVar
A freely accessible database that aggregates information on human genetic variants and their clinical significance. It collects reports from various submitters, including clinical labs and researchers, providing classifications of variants along with supporting evidence. Variants are mapped to reference sequences and follow the HGVS naming standard. One can search by gene or variant to view submissions from clinical labs and interpretations.29
Conclusion
Understanding gene structure, function, inheritance patterns, and mutation types is fundamental to diagnosing and managing genodermatoses. Similarly, pedigree analysis and basic genetic epidemiology can help us in patient counselling. As genetic technologies evolve, so must our interpretative skills. Bridging clinical dermatology with molecular genetics is key to personalized care in inherited skin disorders.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Primer on medical genomics part II: Background principles and methods in molecular genetics. Mayo Clin Proc. 2002;77:785-808.
- [CrossRef] [PubMed] [Google Scholar]
- Genomics basics: DNA structure, gene expression, cloning, genetic mapping, and molecular tests. Semin Cardiothorac Vasc Anesth. 2006;10:282-90.
- [CrossRef] [PubMed] [Google Scholar]
- Teaching molecular genetics: Chapter 1--background principles and methods of molecular biology. Pediatr Nephrol. 2006;21:169-76.
- [CrossRef] [PubMed] [Google Scholar]
- Clarifying mendelian vs non-mendelian inheritance. Genetics. 2024;227:iyae078.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- From Mendel’s laws to non-Mendelian inheritance. Nat Rev Genet. 2024;25:677.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Mechanisms of non-mendelian inheritance in genetic disease. Hum Mol Genet. 2004;13 Spec No 2:R225-33.
- [CrossRef] [PubMed] [Google Scholar]
- Incomplete penetrance and variable expressivity: Is there a microRNA connection? Bioessays. 2009;31:981-92.
- [CrossRef] [PubMed] [Google Scholar]
- Phenotype Variability: Penetrance and Expressivity | Learn Science at Scitable. Available from: http://www.nature.com/scitable/topicpage/phenotype-variability-penetrance-and-expressivity-573 [Last accessed on 2025 Mar 4].
- Cutaneous mosaicisms: Concepts, patterns and classifications. An Bras Dermatol. 2013;88:507-17.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Molecular mechanisms and the significance of synonymous mutations. Biomolecules. 2024;14:132.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Key concepts in genetic epidemiology. Methods Mol Biol. 2018;1793:7-24.
- [CrossRef] [PubMed] [Google Scholar]
- A review of next-generation genetic testing for the dermatologist. Pediatr Dermatol. 2013;30:401-8.
- [CrossRef] [PubMed] [Google Scholar]
- Next-generation sequencing in diagnostic pathology. Pathobiology. 2017;84:292-305.
- [CrossRef] [PubMed] [Google Scholar]
- Practice resource-focused revision: Standardized pedigree nomenclature update centered on sex and gender inclusivity: A practice resource of the national society of genetic counselors. J Genet Couns. 2022;31:1238-4.
- [CrossRef] [PubMed] [Google Scholar]
- Standardized human pedigree nomenclature: Update and assessment of the recommendations of the national society of genetic counselors. J Genet Couns. 2008;17:424-33.
- [CrossRef] [PubMed] [Google Scholar]
- Applied human genetic epidemiology. Adv Exp Med Biol. 2018;1082:145-216.
- [CrossRef] [PubMed] [Google Scholar]
- A clinician’s guide to omics resources in dermatology. Clin Exp Dermatol. 2022;47:858-66.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- OMIM.org: Leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res. 2019;47:D1038-43.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- OMIM.org: Online mendelian inheritance in man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789-98.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002;30:52-5.
- [CrossRef] [PubMed] [Google Scholar]
- Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, eds. GeneReviews®. Seattle (WA): University of Washington, Seattle; 1993. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1116/ [Last accessed on 2025 Jan 24]
- Orphanet. Available from: https://www.orpha.net/. Last accessed on 2025 Jan 23.
- Human gene mutation database (HGMD): 2003 update. Hum Mutat. 2003;21:577-81.
- [CrossRef] [PubMed] [Google Scholar]
- HGMD® home page. Available from: https://www.hgmd.cf.ac.uk/ac/index.php [Last accessed on 2025 Jan 23].
- ClinVar. Available from: https://www.ncbi.nlm.nih.gov/clinvar/ [Last accessed on 2025 J 23].




