
Translate this page into:
Genetics for dermatologists. Part 2: Clinical evaluation, sequencing technologies and interpretation

Corresponding author: Dr. Divya Gupta, Department of Dermatology, Dr BR Ambedkar Medical College, Bengaluru, India. divya_gupta@ymail.com
-
Received: ,
Accepted: ,
How to cite this article: Gupta D, Jose TG, Vishwanathan GB. Genetics for dermatologists. Part 2: Clinical evaluation, sequencing technologies and interpretation. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_500_2024
Abstract
Genomic sequencing technologies have revolutionised the diagnostic approach to genodermatoses, facilitating precise diagnosis, offering unparalleled insights into pathogenesis, and guiding personalised treatment strategies. In this article, we provide an outline for clinical evaluation, how to choose the most suitable genetic test for different dermatological presentations and examine indications for single-gene tests, gene panels, whole exome, and whole genome sequencing. We then delve into sequencing technologies, including next-generation sequencing (NGS) and its various platforms, detailing their strengths, limitations, and clinical applications. By understanding these technologies, clinicians will be better equipped to interpret test results accurately and collaborate with genetic counsellors and laboratory experts effectively.
Keywords
Genodermatoses
next-generation sequencing
sanger sequencing
variant classification

Introduction
Genodermatoses comprise all inherited skin disorders, of which single-gene disorders comprise the largest group. The advent of next-generation sequencing (NGS) marked a significant breakthrough in their diagnosis, having become a routine practice in clinical settings.1,2 Many of us lack in-depth knowledge of mutation detection technologies and the interpretation of reported findings. Recognising this gap, this second part of our review builds on the foundational concepts covered in Part 1, moving from basic genetic terminology to clinical evaluation of genodermatoses and the practical application of genetic testing in dermatology. It focuses on enhancing dermatologists’ understanding of sequencing methodologies and interpretation of genetic reports, and how this helps in the management of patients and families.3,4
Chromosomal abnormalities and copy number variations (CNVs) account for a small percentage of genodermatoses and are more prevalent in skin tumours. Gross chromosomal abnormalities rarely present solely with dermatological problems and often have prominent multi-system involvement, including mental retardation. Molecular diagnostic techniques like comparative genomic hybridisation (CGH) and DNA microarrays along with cytogenetic techniques like karyotyping and fluorescent-in-situ-hybridisation (FISH) are used to evaluate this group of conditions and have been excluded from the review.
Steps in genetic screening of genodermatoses
History and examination
Dermatologists must be aware of red flag signs, such as erythroderma, collodion baby, blisters on sites of friction (at birth), therapy-resistant eczema with infections and growth failure, palmoplantar keratoderma, photosensitivity, poikiloderma (in early childhood), multiple benign or malignant tumours, cutaneous lesions in a linear/ segmental distribution, multiple hypo- or hyper-melanotic macules, loose hanging skin, positive family history and presence of other extra-cutaneous manifestations involving the central nervous system, skeletal system, teeth, eyes and ears (at any age).1 Salient points in history and examination of genodermatoses are shown in Table 1 and Figures 1a, b.5
| History |
Family history: Pedigree chart noting affected individuals, consanguinity, and patterns of inheritance. If possible, examine first- or second-degree relatives and obtain clinical pictures and medical records; ethnicity Prenatal and neonatal history: recurrent abortions, stillbirths and other birth complications, prematurity, type of delivery, or early skin abnormalities. Skin disease symptoms: blisters, generalised scaling, history of collodion membrane, therapy resistant eczema, photosensitivity, pigmentary changes, multiple tumours, anhidrosis, hypohidrosis, heat intolerance Skin disease onset: Age of onset, progression, triggering factors, and response to treatments. For e.g., age dependent changes in clinical features can occur in many conditions like IP, NF, OCA etc. Systemic features: e.g., seizures, delayed milestones, cardiovascular, pulmonary or renal symptoms |
| General Examination | Evaluate for dysmorphic features, skeletal abnormalities, or growth retardation suggestive of syndromic involvement. |
| Dermatological Examination |
Distribution of lesions: Segmental, generalised, or localised patterns (e.g., segmental NF1). Morphology of lesions: Specific findings such as café-au-lait macules, naevi, lentigines, erythroderma, bullae, erosions, scarring, milia, ichthyosis, pigmentary abnormalities, poikiloderma, palmoplantar keratoderma, freckles, photosensitivity, skin tumours, cutis laxa, adermatoglyphia, capillary or arterio-venous malformations etc Hair examination: alopecia, brittle hair, sparse hair, poor hair growth, easy breakability (e.g., pili torti in Menkes syndrome), atrichia Nail changes: Pitting, thickening, dystrophy (e.g., in ectodermal dysplasia), anonychia (EB), hypoplastic nails Mucosal examination: Oral, ocular, or genital involvement (e.g., oral leukokeratosis in pachyonychia congenita; oral lentigines in Peutz Jeghers syndrome; angioid streaks in PXE; heterochromia irides in WS, nystagmus in OCA; oral, conjunctival and corneal erosions in EB; Lisch nodules/ optic glioma in NF |
| Assessment of extra-cutaneous features |
Neurological assessment: e.g., seizures, hypotonia, or neurodevelopmental delays in various genodermatoses Cardiac and vascular anomalies: e.g. arrhythmias, cardiomyopathy (Naxos or Carvajal syndrome); aortic dilatation and dissection in Marfan’s syndrome; or vascular malformations (e.g., Sturge-Weber syndrome). Pulmonary: e.g. lymphangioleiomyomatosis in TSC; Renal/ liver/ GIT: benign renal angiomyolipomas, epithelial cysts, oncocytoma, renal cell carcinoma in TSC; mucocutaneous telangiectasias including GIT in HHT; various tumour syndromes Skeletal: e.g. sphenoid wing dysplasia, pseudoarthrosis in NF; skeletal abnormalities in EDS/ Marfan’s syndrome Immune system: recurrent bacterial or viral infections, bleeding tendencies (Wiskott Aldrich syndrome; Chediak Higashi syndrome), hemophagocytic lymphohistiocytosis (GS- type 2) Hearing – e.g., sensorineural hearing loss (WS), accessory tragi Dental anomalies: e.g. ectodermal dysplasia or amelogenesis imperfecta features; conical teeth in Gardner’s syndrome; dental caries and enamel loss in EB; dental enamel pits and intraoral fibromas in TSC |
| Specialised dermatological investigations |
Skin biopsy: Histopathological and immunohistochemical evaluation with special stains. For e.g., multiple sebaceous skin tumours in Muir-Torre syndrome; trichilemmomas in Cowden’s syndrome; trichoepitheliomas in Brooke-Spiegler syndrome Hair shaft microscopy: Identification of specific patterns (e.g., tiger tail in trichothiodystrophy; bamboo hairs or golf tee hairs in Netherton syndrome); Examine eyebrow and eyelash morphology also Wood’s lamp examination: Highlight depigmentation or pigmentation changes. Dermoscopy and trichoscopy – e.g. skin tumours; confirm findings of hair shaft microscopy Electron microscopy – in EB, EDS Immunofluorescence antigen mapping- EB |
| Other investigations and documentation |
Biochemical tests and enzyme assays. For e.g., to rule out inborn errors of metabolism Radiographic imaging -if skeletal involvement is suspected Barium swallow in EB USG, CT, and MRI (for e.g., neurological involvement in NF or TSC; pheochromocytoma in NF) High-resolution clinical photographs for documentation. |
IP: Incontinentia pigmenti, NF: Neurofibromatosis, OCA: Oculocutaneous albinism, EB: Epidermolysis bullosa, PXE: Pseudoxanthoma elasticum, WS: Wardenburg syndrome, TSC: Tuberous sclerosis, HHT: Hereditary haemorrhagic telangiectasia, GS: Griscelli syndrome, EDS: Ehlers Danlos syndrome, TSC: Tuberous sclerosis, USG: Ultrasound, CT: Computed tomography, MRI: Magnetic resonance imaging, GIT: Gastrointestinal tract
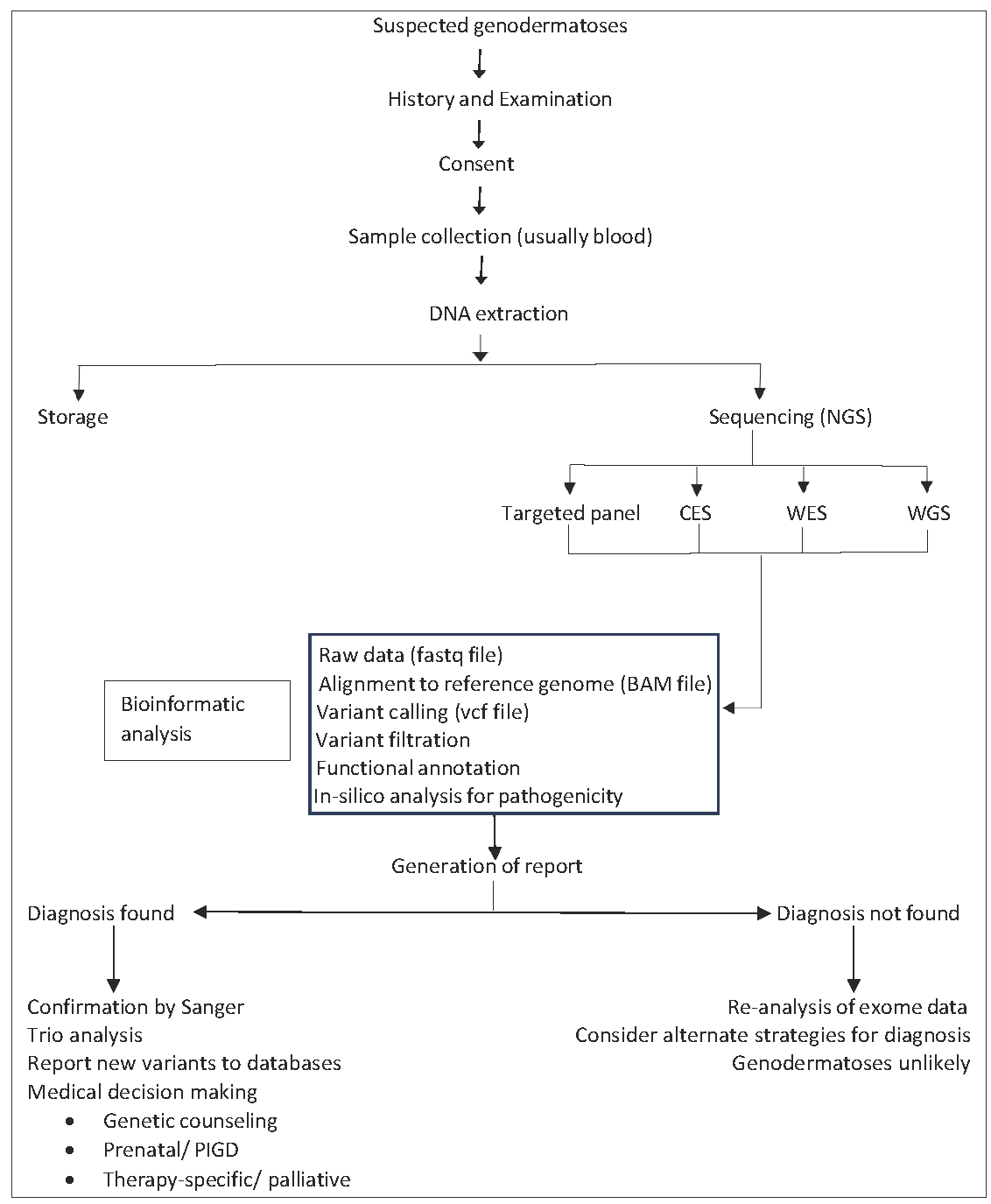
- Work-flow of a suspected case of genodermatosis. (NGS: Next generation sequencing, CES: Clinical exome sequencing, WES: Whole exome sequencing, WGS: Whole genome sequencing, PIGD: Pre-implantation genetic diagnosis)
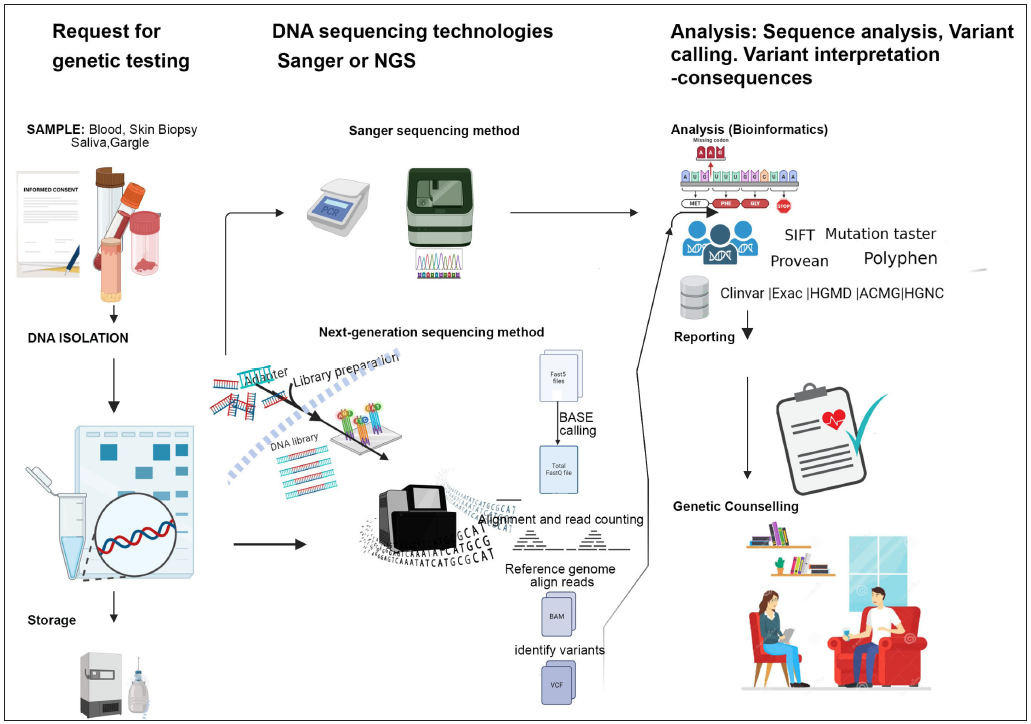
- Pictorial representation of the work-flow of a suspected case of genodermatosis. (NGS: Next generation sequencing, HGMD: Human Gene Mutation Database, ACMG: American College of Medical Genetics and Genomics, HGNC: HUGO Gene Nomenclature Committee, SIFT: Sorting Intolerant from Tolerant).
Pedigree charting is crucial in the history and examination of genodermatoses because it helps identify patterns of inheritance, such as autosomal dominant, autosomal recessive, or X-linked traits. By documenting the family history of affected individuals, clinicians can pinpoint potential genetic links, assess the risk of recurrence in future generations, and guide genetic testing and counselling.
The approach to genodermatoses may be ‘phenotype first’ or ‘genotype first’. A better approach is ‘phenotype first’. Here, the clinician considers medical and family history to understand patterns of inheritance. The clinician also performs examination, and does relevant screening investigations - all of which help to arrive at phenotype. This gives us possible clinical differential diagnoses, following which one can proceed for genotyping. It is better to analyse the clinical phenotype before testing for genotypes.
However, with increasing ease of access to sequencing technologies, it is often tempting to order a genetic test when one suspects a genetic problem without completely phenotyping the patient. We also see patients in our OPD directly walking in with a genetic report. This approach of doing genetic testing before phenotyping is called ‘genotype first’ and can be disadvantageous on many accounts – 1) genetic test may come negative (the yield of whole exome sequencing, the most commonly ordered test for genodermatoses, based on a genotype first approach, is only 50%)5 2) it may throw up a variant of uncertain significance (VOUS) and good phenotyping is essential to narrow down the variants 3) phenotype is important for initiating therapy, which cannot be decided by the genotype alone 4) Phenotype first approach also helps us to select the right genetic test.
Hence, good, clinical, and detailed phenotyping and screening investigations are important. However, we often need both for the final diagnosis since it helps in genetic counselling and offering precision therapy.5
Most genodermatoses run in families, and the absence of a family history does not exclude a genetic skin disorder.
Factors influencing diagnosis may include asymptomatic female carriers of X-linked disorders, early death, incomplete family history, and false paternity.1 Non-mendelian inheritance patterns that do not follow the classic Mendelian rules also add complexity to genetic inheritance. Indications for genetic testing are enumerated in Table 2.1,2
| Patients with unclear phenotypes |
| Determination of prognosis or anticipated medical issues |
| Initiation of specific therapy if available/early intervention |
| Identifying carriers and segregation of mutation in the family |
| For performing prenatal diagnosis/pre-implantation genetic testing |
Counselling
Pre-test counselling
A genetic test is extensive and complicated, and requires families to undergo a prior consenting session. This crucial step educates the parents and patients about sample type (usually blood), the selection of family members for sample collection (proband vs. trio vs. other), turnaround time, test cost, possible results (including the possibility of finding variants of uncertain significance (VOUS)) and incidental findings unrelated to the suspected disease, and associated limitations and risks ( for e.g., privacy concerns and how the results could affect other family members). Many families believe that a genetic test will lead the way to a cure or treatment for their child. It must be emphasised to them that this isn’t guaranteed.2 At present, genetic tests continue to be expensive and out of reach for most patients. They are often not covered under insurance. This may change in the future as the identification of patients with genodermatoses increases and specific treatments are developed.4,6
Post-test counselling
This is performed after the clinician receives the results of genetic testing. These are discussed with the patient or family, including any identified variants and their clinical significance, if known. This session is crucial to help them understand the results, whether they are diagnostic, inconclusive, or require further testing. Patients are guided on the next steps for management or follow-up, and if no clear diagnosis is found, the possibility of future re-evaluation as more information becomes available is also discussed.
Sample collection
Sample collection, labelling, storage, and transportation are crucial steps, necessitating that they are done correctly [Table 3].7,8
| Specimens |
Peripheral blood in EDTA (purple top) containing tube – 2-4 ml, minimum 1 ml (heparin must be avoided). Saliva - kit based collection, minimum 1 ml ⴕ Buccal swabs, minimum 3 swabs ⴕ Cultured cells, minimum 2 x T25 flasks (confluent) Formalin-fixed paraffin-embedded specimens from histopathology€ Forensic samples |
| Sample collection |
Collect samples as mentioned above Wrap it with a cling wrap/ micropore tape to prevent spills Label with name, age, gender, date of birth, date of collection Do not label family members’ specimens with the proband’s name |
| Completion of paperwork |
Fill the consent forms. Complete the clinical notes and pedigree chart, including the physician’s signature* Mention if the patient wishes to opt out of receiving medically actionable secondary/ incidental findings and DNA storage after completion of the test. |
| Transport of sample |
Place collection tubes in a plastic/ cardboard box containing cotton or bubble wrap and pack it well** Place the cardboard box along with the clinician’s notes in a courier package Label the address and phone number of the receiver correctly and post; Ideally, the sample must be shipped on the day of collection and reach the analysis facility in 24-48 hours. In case longer transit times are expected, then a refrigerated gel pack/ ice pack must be included in the box. Germline DNA extraction must be complete within 48-72 hours of blood draw. |
| Storage of specimen# |
At 4°C, blood specimens stable for up to 1 week Frozen blood specimen is stable for up to 1 month Extracted DNA can be stored at -80°C for long-term |
ⴕ Provide a non-invasive alternative to venesection, but at the expense of quantity and quality, especially DNA contamination from oral microflora and food remnants
€Risk of poorer DNA quality
#Storage of blood can be considered when sequencing is not possible immediately.
Sequencing and its types
Sequencing is a chemical process that establishes the order in which four nucleotides (Adenosine, Thymine, Cytosine, and Guanine) are chained into a DNA strand. Broadly, for diagnostic purposes, there are 2 types of sequencing:
Sanger sequencing (SS) (also known as first-generation sequencing)– It is the traditional and low throughput method of genetic testing. It is time-consuming (can take upto 3-7 days per gene, depending on the length of the exon), laborious, and expensive, allowing the sequencing of only one gene at a time. However, the read lengths in SS are relatively longer, capturing 800-1000 base pairs of nucleotides with a low error rate and high accuracy. The data generated by Sanger sequencing are relatively straightforward to interpret. Thus, despite its slower speed and higher cost per base compared to NGS, SS has always been the gold standard to detect or confirm pathogenic single nucleotide variants (SNVs). It can be an effective tool for sequencing small, specific regions such as individual genes or exons.9 Clinical settings in which SS is used include i) identifying single gene disorders ii) confirmatory testing to validate the variants identified by NGS iii) prenatal testing to analyse foetal DNA for known familial mutations before birth iv) carrier screening in other family members of a confirmed case
Next-generation sequencing (NGS) (also known as second-generation sequencing/massive parallel sequencing/high-throughput sequencing): The completion of the reference human genome in the 2000s, along with the evolution in sequencing chemistry marked a watershed moment for genetic technology as fast and high-speed sequencers were made available for high-throughput sequencing, thus enabling simultaneous sequencing of hundreds of genes. NGS is faster, cost-effective, has better diagnostic accuracy, and is currently the method of choice for identifying pathogenic mutations. The steps of NGS are depicted in Figure 2.
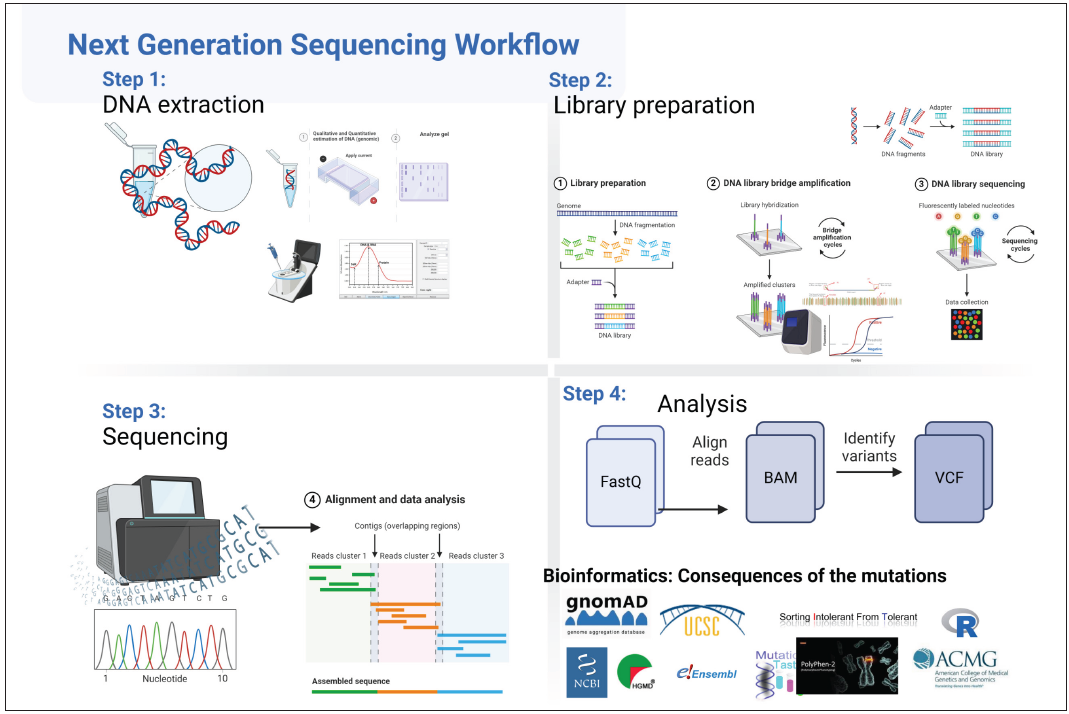
- Steps in next generation sequencing Step1: DNA extraction: DNA is extracted from blood through traditional or commercially available kits. Quantification of the nucleic acid is done (Quality and concentration are noted). Step 2: Library preparation: Fragmentation of DNA is done by diverse methods like sonication/mechanical/ enzymatic etc. followed by hybridisation capture or amplicon assay for target regions, leading to library preparation. Step 3: Sequencing: The DNA library is loaded onto sequencer and massive parallel sequencing of multiple individually amplified fragments takes place. Step 4: Bioinformatic Analysis The above steps result in generation of raw data (FastQ files) which undergo the following steps: Quality control→ Adaptors removal→ Alignment to the reference genome→ Variant calling (or variant discovery) to call out specific mutants→ Variant filtration to filter in variants of interest→ Functional annotation and interpretation to infer the biological and clinical role (last two steps together known as Filtering and Annotation). Simply put, the last step compares patient’s nucleotide sequences with those of healthy individuals’ databases. Variants in the patient’s DNA differing from those in the controls are identified through this analysis. These annotated variants are then evaluated for pathogenicity using various computer programs that consider the mutations’ effects on protein function or predict their impact on splicing.
Types of next-generation sequencing
Clinical applications of NGS include targeted panel sequencing, clinical exome sequencing (CES), whole-exome sequencing (WES), whole-genome sequencing (WGS), and RNA-sequencing. These are compared in Table 4.2,3,10–12
| Targeted panel sequencing | Clinical exome sequencing (CES) | Whole exome sequencing (WES) | Whole genome sequencing (WGS) | |
|---|---|---|---|---|
| Definition | Gene panels for specific diseases. E.g., EB gene panel covers 17 genes known to be associated with EB and other skin fragility syndromes only17 | Sequencing of only those genes in the coding region that are known to be associated with various Mendelian disorders | Sequencing of complete coding regions (exons) of the genome | Sequencing of coding and non-coding (exons + introns) regions of the genome |
| Indication | When the disease is very well characterised phenotypically, E.g., Ichthyosis or XP panel | Similar to WES. |
High suspicion for a genetic disorder without a clear candidate syndrome; For phenotypes where there is significant genetic heterogeneity; Where more specific genetic testing has failed to yield a diagnosis Currently mainstream application worldwide |
There is a strong suspicion of an inherited Mendelian disorder, and other genetic testing (E.g., CES, WES, karyotyping, chromosomal microarray) was inconclusive. Can increase diagnosis rates by 10-15%* |
| Coverage | Covers the specified panel of genes | Covers ⁓ 25% of the exome (5000 genes) and is continuously expanding | Covers 1-3% of the genome |
Detects 95% of the genome Mutations in the noncoding regions can be detected for e.g. micro-RNA, promoter regions, and ultraconserved elements, some of which are critical for gene expression or DNA stability. |
| Cost | Targeted panel sequencing < CES < WES <WGS | Targeted panel sequencing < CES < WES <WGS |
Targeted panel sequencing < CES < WES <WGS Increasingly however, its price has decreased enough that it is now a viable option in many clinical scenarios, particularly when a broad genetic assessment is required. |
Targeted panel sequencing < CES < WES <WGS |
| TAT | Targeted panel sequencing < CES < WES < WGS | Targeted panel sequencing < CES < WES < WGS | Targeted panel sequencing< CES < WES <WGS | Targeted panel sequencing < CES < WES <WGS |
| Dataset size | Small dataset, easy to analyse and interpret. VOUS generation is limited to the panel of genes being tested | While larger than a Targeted Panel, a Clinical Exome still avoids generation of a large number of VOUSs (as compared to WES or WGS), making genetic counselling and trio analysis less complex | Datasets are larger than CES with still a large number of VOUSs so difficult to analyse and interpret. | Largest dataset with generation of large numbers of VOUSs; extremely difficult to analyse and interpret (more than CES or WES); currently used in research setting |
| Disadvantages and limitations |
With continuous discovery of new genes, the targeted gene panels often become outdated very quickly. If this has not been updated in the laboratory databases, then test may return a negative With falling costs, likely to be replaced by WES in the future |
Can miss mutations in the rest of the coding and non-coding regions. Difficult to detect structural variants, e.g., small and large deletions/ insertions/translocations/ duplications/ |
Similar to CES Does not detect mutations in non-coding regions, especially, in regulatory, promoter, and ultra-conserved regions of DNA. Difficult to detect structural variants like small and large deletions/ insertions / translocations/ duplications/ CNVs Cannot detect synonymous or silent nucleotide substitutions in exons Useful for SNVs and indels but less useful for CNVs |
Some ability to detect structural rearrangements and CNVs- insertion or deletion events (plus or minus up to seven base pairs) can be detected with an accuracy of 80%, and larger insertion or deletion events are detected with less accuracy. Useful for CNV although less useful for SNV and small insertion/deletion (indels) |
CES: Clinical exome sequencing, WES: Whole exome sequencing, WGS: Whole genome sequencing, EB: Epidermolysis bullosa, XP: Xeroderma pigmentosum, NGS: Next generation sequencing, VOUS: Variant of uncertain significance, TAT: Turn-around time, SNV: Single nucleotide variant, CNVs: Copy number variant
Important points to remember:
-
Genetic testing should be done at certified labs with clear protocols for lab audits. For commercial labs doing NGS, it is good to have CAP (College of American Pathologists) and NABL (National Accreditation Board for Testing and Calibration Laboratories) accreditation, too, as it ensures internationally recognised standards of quality. Results must be interpreted by a qualified clinical molecular geneticist or molecular genetic pathologist. If the lab is doing prenatal genetic tests, then it must possess a PCPNDT (Pre-Conception Pre-Natal Diagnostic Technique) certificate. Sometimes, the decision of research labs actively involved in NGS, to not pursue accreditation stems from their unique operational needs.
-
The physician who orders the genetic test must have sufficient knowledge of genetic mechanisms to order and correctly interpret the results.
-
Turnaround time varies between 4-12 weeks, depending on the type of test ordered. The clinician needs to take this into account while ordering prenatal tests, as the window period for intervention, in case the foetus is inviable, is only up to 20 weeks.
-
Clinical exome sequencing (CES) or Whole exome sequencing (WES) is preferably carried out in trio testing (mother, father, affected child), as it enhances the diagnostic yield compared with singleton analyses. This approach helps identify and interpret de-novo and compound heterozygous variations more easily. Trio testing is easier to perform in paediatric patients as compared to affected adults. In the absence of parents’ sample, the inclusion of siblings can increase the ability to diagnose recessive disorders.2 Another cost-effective approach is to analyse the affected child sample by NGS followed by carrier screening by the Sanger method in parents or siblings.
-
Depth and Coverage - In exome sequencing, “depth” and “coverage” refer to two important metrics that assess the quality and completeness of the sequencing data:
-
Depth (also known as sequencing depth or read depth)- refers to the average number of times each base in the target regions of the exome are sequenced. It indicates how many times a specific nucleotide has been read during the sequencing process. A higher depth means more confidence in the accuracy of the sequencing results for that base. For example, if a target region has a depth of 20x, it means that, on average, each base in that region has been sequenced 20 times.
-
Coverage- refers to the proportion of target regions in the exome that has been sequenced at a certain depth. It represents the completeness of the sequencing data across the regions of interest. For example, if a target region has 1000 base pairs, and the sequencing has covered only 900 base pairs, it means that the coverage is 90%.
-
Both depth and coverage are essential for ensuring the accuracy and reliability of exome sequencing results. Higher depth and coverage generally lead to better detection of genetic variants. Insufficient depth or coverage can result in missed variants or inaccuracies in variant calling. For germline variants, a depth of 80-100x is desirable, whereas for somatic variants, with low variant frequency, deep sequencing (upto 250x) is desirable.
-
Targeted approaches may require higher depth and coverage in specific regions of interest, whereas broader sequencing approaches like WES and Whole genome sequencing (WGS) aim for comprehensive coverage of exonic or genomic regions with possibly a lower but optimal depth to achieve reliable variant detection.
-
Standard NGS has challenges, with some regions of the genome being poorly covered. Large genes, pseudogenes, specific regions with high GC (guanine-cytosine) content, small indels (small insertions and/ or deletions of nucleotides), and trinucleotide repeats are challenging to sequence and interpret, leading to a coverage of <100%. High GC content can make DNA regions difficult to sequence because the strong bonds between G and C bases are harder for sequencing machines to read accurately. These regions may have gaps or lower-quality data compared to areas with more balanced base content.
-
There may be differences in coverage between different sequencing platforms (for e.g., Illumina vs Ion Torrent), leading to differences in results between labs. Illumina is highly accurate, particularly for detecting SNVs, whereas Ion Torrent has a rapid turnaround time but higher error rates.
-
Sequencing errors are also common, therefore, validation of NGS data using Sanger sequencing is desirable.13
-
We still do not understand large parts of the coding region (exome), hence it is difficult to interpret the results of sequencing. Pathogenic variants may be missed during variant filtration and variant calling. Moreover, predicting the pathogenic effect of novel variants/ variants of uncertain significance (VOUS/ VUS) is difficult despite prediction from in-silico tools.
-
The overall approximate diagnostic rate for genodermatoses is around 50%. The diagnostic rates are higher and approach 95-100% for disorders that are phenotypically well characterised and nearly exclusively monogenic, such as epidermolysis bullosa. In such cases, small, targeted panels can be used as first-line diagnostic test.5
-
In patients suspected for genetic disorders where DNA sequencing does not reveal any significant mutations, RNA-sequencing (RNA-seq) is emerging as a complementary method. Unlike DNA-based approaches, RNA-seq directly evaluates RNA sequences and can identify aberrant transcripts. For e.g., in a patient with splice site mutations, clinicians can identify abnormal splicing events, such as exon skipping or intron retention which are not obvious in DNA sequencing.
-
Storage of vast amounts of data generated from NGS studies poses a significant challenge. Issues like hardware limitations, patient confidentiality, and data accessibility remain pressing and unsolved.14,15
Reasons for non-contributory WES/ No variants found in the report:
-
Cannot detect non-coding variants as the targeted regions include all protein-coding exons and approximately ±20 base pairs from the exon-intron boundary.9
-
Can miss structural variants – insertions, deletions, duplications, inversions, translocations, copy number variants, owing to the alignment process.
-
Variants may be interpreted differently among clinical laboratories, leading to disagreements in around 10% of cases.3
-
Inadequate clinical data provided by the clinician leading to errors in variant calling and filtration.
In inconclusive cases one must preserve the genetic material along with a detailed description of clinical features and images. Many times, the responsible gene may be found in future analyses. Regular follow-up visits will help unearth new clinical features with time, eventually leading to a diagnosis.1
Re-analysis of exome data, providing complete and correct phenotype to enable variant filtering at the time of doing bioinformatics, and consultation between the clinician and the lab will increase the diagnostic yield.
How to read and understand a genetic report?
Ideally, an interdisciplinary approach involving a team of dermatologists, clinical geneticists, and laboratory experts should be followed for accurately interpreting genetic reports in genodermatoses, as it ensures a thorough understanding of the genetic findings in the context of the patient’s clinical presentation. Dermatologists provide clinical context, correlating test results with skin findings and history, while clinical geneticists discuss inheritance patterns, variant classification, and counselling. Laboratory experts ensure accurate analysis and interpretation of sequencing data considering the information provided by the clinicians. This team-based approach is critical, especially in complex genodermatoses.
The components of an NGS report are mandated as per American College of Medical Genetics and Genomics (ACMG) guidelines.6 These are as follows:
Results
This includes a list of any candidate variants that contribute to the patient’s phenotype using the Human Genome Variation Society (HGVS) nomenclature (see below). Variants are presented in tabular form with - nomenclature at both the nucleotide (genomic and cDNA) and protein levels. The order of reporting is; gene name- exon number- coding position/ protein position- zygosity-disease- inheritance- classification.
Interpretation
The interpretation covers the summary of the available information about each variant i.e.,
-
i.
evidence supporting variant classification (see below),
-
ii.
predicted impact on the proteins, including functional data from in-silico (see below) and evolutionary conservation analyses.
-
iii.
whether the variant has been reported before.
-
iv.
whether identified variants explain the patient’s indication for testing and
-
v.
recommendations for supplemental clinical testing (e.g., enzymatic/functional testing of the patient’s cells and variant testing of family members).
Aspects like decreased penetrance and variable expressivity of the disorder can be discussed in the final report, if applicable.
References
The references, if any, that contributed to the ACMG classification are listed at the end of the report.
A brief overview of different aspects of ACMG guidelines relevant for clinicians:
Nomenclature
The Human Genome Variation Society (HGVS) maintains standard gene variant nomenclature available, at (http://www.hgvs.org/mutnomen). It is recommended as the primary guideline for variant nomenclature, with laboratories specifying the version used in their test methods. Tools (https://mutalyzer.nl) can be used to generate accurate HGVS nomenclature for variant descriptions. E.g., according to HGVS, nonsense variants are reported as “*” or “Ter”, or even “X”.16
Terminology and ACMG variant classification
Frequently used terms like “mutation” and “polymorphism” often lead to confusion due to incorrect assumptions of pathogenic and benign effects, respectively. Hence, it is recommended to replace both terms with “variant/variation”. Since the clinical significance of a variant can span a spectrum from pathogenic to benign, the ACMG guidelines prescribe standardised terms like ‘pathogenic,’ ‘likely pathogenic,’ ‘uncertain significance,’ ‘likely benign,’ and ‘benign’ for further classification. Broad criteria on which this classification is based are depicted in Table 5.
| Type of variant – for e.g., termination mutations are usually pathogenic or more severe. Certain types of mutations (e.g., nonsense, frameshift, canonical +/−1 or 2 splice sites, initiation codon, single exon or multi-exon deletion) typically disrupt gene function by causing complete absence of the gene product through lack of transcription or nonsense-mediated decay of an altered transcript. |
| Position (mutations in - known ‘hot-spot’ region; crucial site for protein function; present in an evolutionarily conserved site - are likely to be pathogenic; variants in intronic regions are likely to be benign) |
| Inheritance |
| Segregation |
| Prevalence (in healthy and diseases individuals) - Variant frequency of >5% in the general population is predictive of benign nature |
| Functional data relevant to gene function e.g., experiments in animal models may demonstrate pathogenic effects of the variation. |
| In-silico predictions (Prediction scores from software tools may favour pathogenicity) |
| Alternate locus observations- In some conditions, having multiple variants can worsen or improve the disease phenotype. |
|
Variant spectrum- If a gene is known to cause a disease when it has missense variants, finding similar missense variants in that gene is likely to mean that it is harmful. On the other hand, if a gene causes disease only when large parts are missing (truncating variants), then missense variants in that gene are probably harmless. This pattern can be used to decide whether a new genetic change is likely to be harmful or not. |
| Same amino acid change- If a particular missense variant is known to be pathogenic, another nucleotide change leading to the same amino acid can also be considered pathogenic in most cases. |
Variant of uncertain significance (VOUS/VUS)
In many cases, a variant’s pathogenic or benign status isn’t convincingly established, leading to its classification as a VOUS or VUS. A VOUS is uncertain; it might cause a disorder or be entirely benign, but insufficient data hinders definite determination. Functional validation in cell culture or animal models, beyond current clinical evaluation, is needed for VOUS. While every exome harbours dozens of VOUSs, only those linked to the patient’s phenotype are reported. It must be emphasised that it shouldn’t guide clinical decisions, and efforts should focus on determining the variant as “pathogenic” or “benign.” Over time, increased genetic data sharing, as well as collaboration between clinical and research labs will reclassify more VOUSs. Laboratories must suggest periodic inquiries by healthcare providers to stay updated on any changes in knowledge regarding VOUSs.
When a clinician encounters a genetic report with a VOUS, the following approach is recommended:
-
Review clinical context: Correlate the patient’s clinical presentation with the variant in question and check the family history and inheritance pattern of the disorder.
-
Evaluate parental testing: If an asymptomatic parent carries the same VOUS, it might suggest that the variant is less likely to be pathogenic, especially for autosomal dominant conditions. Other family members can be tested to see if the variant co-segregates with the disease phenotype.
-
Population frequency: If the VOUS has a high frequency in the general population (>5%), it is less likely to be pathogenic, particularly for severe or rare conditions. Use databases such as gnomAD to check variant frequency.
-
Functional studies: Look for existing in vitro or in vivo functional studies on the variant. If none exist, consider collaborating with research institutions to investigate its impact on protein function.
-
In silico predictions: Utilise bioinformatics tools that predict the potential impact of the variant on the protein structure and function, though these should be interpreted with caution.
-
Literature review: Search for published reports that describe the variant and its association with disease.
-
Consultation with genetics professionals: Engage with genetic counsellors or clinical geneticists to interpret VOUS and determine next steps for patient management.
-
Reclassification over time: Periodically re-evaluate the VOUS as new data becomes available. Genetic databases are continuously updated, which might lead to the reclassification of the variant from uncertain significance to being either benign or pathogenic in the future.
-
Patient communication: Clearly communicate the uncertainty of the VOUS to the patient and discuss the potential for future reclassification.
-
Management plan: Establish a plan that may include regular monitoring and follow-up, especially if the patient is at risk for developing symptoms.
Secondary/incidental findings
Secondary or incidental findings are medically actionable genetic variants discovered during genetic testing that are unrelated to the initial purpose of testing. The ACMG has identified 73 genes with pathogenic variants associated with an increased risk of cancer or sudden death, offering medical interventions like prophylactic mastectomy or pacemaker implantation.3 However, these interventions carry medical and psychological risks. Counselling before testing involves informing individuals about the chance of secondary findings and the option to opt out of receiving such results in the consent form.
In-silico software tools
Various in-silico tools or software algorithms, available publicly and commercially, assist in interpreting the effect of sequence variants at nucleotide, protein and amino acid levels. Combining predictions from different tools enhances interpretation robustness. In-silico tools, such as PolyPhen2, SIFT, and MutationTaster, are commonly used in clinical labs for missense variant interpretations. A list of variant prediction in-silico tools can be found in Table 6.
| Missense variants18-22 |
| PolyPhen-2 - Predicts the effect of single amino acid substitution on protein’s structure and function.http://genetics.bwh.harvard.edu/pph2/ |
| SIFT- It sorts out the intolerant from the tolerant and predicts the impact of amino acid substitution https://sift.bii.a-star.edu.sg/ |
| Mutation Taster- Prediction tool that measures the effect of the DNA variants. https://www.mutationtaster.org/ |
| CADD- ML tool that predicts the functional effects of the DNA variants. https://cadd.gs.washington.edu/snv |
| Mutation Assessor- Predicts the impact of amino-acid substitutions functionally. http://mutationassessor.org/r3/ |
| Splice site (SS) variants23,24 |
| NNSplice-predicts the splice sites by neural networks https://www.fruitfly.org/seq_tools/splice.html |
| GeneSplicer-Computational method which detects the splice-sites https://ccb.jhu.edu/software/genesplicer/ |
| Insertion-Deletion (Indels) variants20,25,26 |
| PROVEAN - Tool that predicts whether the SNV or Indel has functional impact https://www.jcvi.org/research/provean |
| Mutation Taster- Prediction tool that measures the effect of the DNA variants. https://www.mutationtaster.org/ |
| MutPred-Indel- Web application tool to detect indels. http://mutpred2.mutdb.org/mutpredindel/ |
| Frameshift and stop gain variants27–29 |
| MutPred-LOF- Web application tool to detect frameshift and stop gain variants. http://mutpred2.mutdb.org/mutpredlof/ |
| DDIG-ML method which detects nonsense variants. https://sparks-lab.org/server/ddig/ |
| LOFTEE- Plugin developed to discern loss of function variants. https://github.com/konradjk/loftee |
PolyPhen-2: Polymorphism Phenotyping v2; SIFT: Sorting Intolerant From Tolerant; CADD: Combined Annotation-Dependent Depletion; ML- machine learning; NNSplice: Neural network based splicing production program; PROVEAN: Protein Variation Effect Analyzer; MutPred-LOF: MutPred loss of function; DDIG: Detecting DIsease-causing Genetic variations; LOFTEE: Loss-Of-Function Transcript Effect Estimator
Databases
Newer discovered variants get continuously added to the databases [Table 7] and serve as useful sources of information for clinicians as well as geneticists. Databases can be of 2 types: Population database and Disease database.
-
i.
Population databases: provide variant frequencies in large populations. These include pathogenic variants as well as healthy individuals. However, they lack comprehensive information on variant functional effects or associated phenotypes.
-
ii.
Disease databases: focus on variants in patients with diseases, along with the assessment of variant pathogenicity.
| Population databases | Disease databases |
|---|---|
| 1000 Genome | ClinVar |
| gnomAD | Ensembl |
| dbSNP | OMIM |
| GenomeAsia | HGMD |
| IndiGenomes | LOVD |
| TOPMed | DECIPHER |
GnomAD: Genome aggregation database, dbSNP: The Single Nucleotide Polymorphism database, TOPMed: Trans-Omics for Precision Medicine, OMIM: Online Mendelian Inheritance in Man, HGMD: Human Gene Mutation Database, LOVD: Leiden Open source Variation Database, DECIPHER: DatabasE of Chromosomal Imbalance and Phenotype in Humans using Ensembl Resources
Special considerations
ACMG guidelines are followed for inherited Mendelian disorders and are not meant for interpretation of somatic variations, pharmacogenomic variants, variants in genes associated with multigenic non-Mendelian complex disorders, or for interpretation of genes of uncertain significance.6
Pharmacogenomics
Some labs offer additional information about variants in genes involved with drug metabolism that affect drug efficacy and confer increased risk for adverse events.
Common complex disorders
Variations in common, complex disease genes are sometimes identified while sequencing Mendelian genes. These are reported as “risk alleles” or under “other reportable” categories in the diagnostic report, instead of using the terms “pathogenic”, and “likely pathogenic”.
Genes of uncertain significance (GUS)
This is when the lab finds variation in a gene that has never been associated with any patient phenotype or if it’s linked to a different phenotype. Variants found in a GUS are reported as ‘Variants in a gene of uncertain significance’. These variants, if reported, should always be classified under “Uncertain Significance”. Further evidence is needed to confirm the gene’s association with a disease before any variant in it is considered pathogenic for that disease.
Conclusion
Genetic tests play an indispensable role in accurate diagnosis, prognosis, and personalised treatment strategies for genodermatoses. As they become increasingly accessible and affordable, it is imperative for clinicians to grasp the fundamentals of ordering genetic tests and interpreting the reports effectively. This review article serves as a comprehensive resource, equipping clinicians with essential knowledge and practical insights to optimise patient care and contribute to the advancement of precision medicine in dermatology.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- A multistep approach to the diagnosis of rare genodermatoses. J Dtsch Dermatol Ges. 2016;14:969-86.
- [Google Scholar]
- A Clinician’s perspective on clinical exome sequencing. Hum Genet. 2016;135:643-54.
- [CrossRef] [PubMed] [Google Scholar]
- Interpretation of genomic sequence variants in heritable skin diseases: A primer for clinicians. J Am Acad Dermatol. 2023;89:569-76.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Access to genetic diagnostics for genodermatoses: Who should get tested? why? who pays? Pediatr Dermatol. 2017;34:105-8.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and molecular diagnosis of genodermatoses: Review and perspectives. J Eur Acad Dermatol Venereol. 2023;37:488-500.
- [CrossRef] [PubMed] [Google Scholar]
- ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15:733-47.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- UCSF Health Center for Clinical Genetics and Genomics. Exome sequencing. Available from: https://genomics.ucsf.edu/content/exome-sequencing. Last accessed Mar 9, 2024.
- Specimen Requirements - PreventionGenetics. Available from: https://www.preventiongenetics.com/ClinicalTesting/specimenRequirements. Last accessed Mar 9, 2024.
- Whole-exome sequencing enables rapid and prenatal diagnosis of inherited skin disorders. BMC Med Genomics. 2023;16:193.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- A review of next-generation genetic testing for the dermatologist. Pediatr Dermatol. 2013;30:401-8.
- [CrossRef] [PubMed] [Google Scholar]
- Whole genome sequencing (WGS), whole exome sequencing (WES) and clinical exome sequencing (CES) in patient care. LaboratoriumsMedizin.. 2014;38:221-30.
- [CrossRef] [Google Scholar]
- Next-generation sequencing in diagnostic pathology. Pathobiology. 2017;84:292-305.
- [CrossRef] [PubMed] [Google Scholar]
- The challenge for the next generation of medical geneticists. Hum Mutat. 2014;35:909-11.
- [CrossRef] [PubMed] [Google Scholar]
- The promise and challenges of next-generation genome sequencing for clinical care. JAMA Intern Med. 2014;174:275-80.
- [CrossRef] [PubMed] [Google Scholar]
- The molecular revolution in cutaneous biology: Era of next-generation sequencing. J Invest Dermatol. 2017;137:e79-e82.
- [CrossRef] [PubMed] [Google Scholar]
- Describing sequence variants. Available from: https://www.hgvs.org/mutnomen/ Last accessed Mar 5, 2024.
- Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-27.
- [CrossRef] [PubMed] [Google Scholar]
- A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812-4.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- MutationTaster2021. Nucleic Acids Res. 2021;49:W446-51.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- CADD v1.7: Using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 2024;52:D1143-54.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011;39:e118.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Improved splice site detection in Genie. J Comput Biol. 1997;4:311-23.
- [CrossRef] [PubMed] [Google Scholar]
- GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185-90.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31:2745-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Pathogenicity and functional impact of non-frameshifting insertion/deletion variation in the human genome. PLoS Comput Biol. 2019;15:e1007112.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- When loss-of-function is loss of function: Assessing mutational signatures and impact of loss-of-function genetic variants. Bioinformatics. 2017;33:i389-98.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- DDIG-in: Detecting disease-causing genetic variations due to frameshifting indels and nonsense mutations employing sequence and structural properties at nucleotide and protein levels. Bioinformatics. 2015;31:1599-606.
- [CrossRef] [PubMed] [Google Scholar]
- The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434-43.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




